基于TCGA和ICGC构建和验证肝细胞癌肿瘤微环境相关基因的预后风险模型
2023-01-15李绍智梁维仁郭金友郑家平
李绍智 梁维仁 郭金友 郑家平
肝细胞癌(hepatocellular carcinoma,HCC)是全球最常见的恶性肿瘤之一,其发病率占恶性肿瘤的第六位,病死率占第四位[1]。近年来有较多治疗方式,包括手术治疗和药物治疗等已经取得巨大进展,但HCC患者的预后仍不理想,其中主要原因是肿瘤的复发和转移[2]。越来越多的研究表明肿瘤微环境(tumor microenvironment,TME)在肿瘤的发生、发展等过程中扮演着重要的角色[3-4]。肿瘤细胞可以通过分泌各种细胞因子、趋化因子等,改变TME并导致周围细胞重新编程,使这些细胞能够促进或抑制肿瘤细胞的生存和发展[3,5]。研究报道TME相关基因在预测不同癌种患者的生存、预后具有重要价值[6-8]。在肝癌上,尽管一些研究已经构建了TME相关基因的预后风险模型,但这些研究缺乏独立数据集的验证,并且预测的性能仍需提高[9-11]。生物信息学方法在探索和鉴定肿瘤预后相关生物标志物上具有重要的价值[12]。本研究基于TCGA和ICGC数据库筛选TME相关的关键基因,构建和验证预后风险评估模型,以期为HCC患者的预后评估提供参考。
1 材料与方法
1.1 数据下载和预处理 HCC转录组数据、临床信息、随访资料和体细胞突变信息从癌症基因图谱(the cancer genome atlas,TCGA)数据库(https://portal.gdc.cancer.gov/)中获取,其中对随访信息有缺失的患者予以删除。其次,独立数据集国际肿瘤基因组协作组(international cancer genome consortium,ICGC)中的HCC队列(LIRIJP)患者的转录组数据也被下载用于验证。根据先前已经发表的TME相关研究[13-19],4,061个TME相关的基因被选择用于后续分析。
1.2 非负矩阵分解算法验证分子亚型 通过单因素Cox分析对TCGA中这4,061个TME基因表达水平进行分析,获得与预后相关的TME基因(P<0.01)。通过非负矩阵分解(non-negative matrix factorization,NMF)对样本进行聚类[20],聚类的数量k根据cophenetic和残差平方和确定。最终选择合适的聚类数量用于后续分析。为了比较不同聚类间微环境细胞群的差异,该方法[19]被用于计算每个样本中各种免疫细胞的评分,包括CD8+T细胞、细胞毒性淋巴细胞、B细胞、中性粒细胞、单核细胞、骨髓树突状细胞等。不同免疫细胞在不同聚类之间的差异也被比较。
1.3 预测模型构建 对临床数据整理过滤后,370例HCC患者(TCGA数据集)以3∶2的比例被随机分为训练组(Training cohort)和测试组(Testing cohort),并比较两组间临床信息以确定是否存在组内差异,进一步通过Lasso、Cox回归寻找合适的预测相关TME基因[21]。Lasso回归能够消除过于相关的基因,防止过度拟合,并能找到误差最小的基因。基于Lasso回归的结果,多因素Cox回归分析被用于进一步筛选重要的与HCC患者预后密切相关的TME枢纽基因。根据TME相关预后基因的表达及其多因素分析产生的回归系数生成最终的预后特征:TME风险评分(TMEScore)=(βmRNA1×mRNA1的表达水平)+(βmRNA2×mRNA2的表达水平)+…+(βmRNAn×mRNAn的表达水平)。TMEScore的cutoff值是根据训练集中的中位TMEScore决定。根据TMEScore的cutoff值,将患者分为高风险组或低风险组。
1.4 预测模型性能评估 基于建立的TMEScore,进一步比较训练集、测试集、整个人群以及独立数据集(ICGC-JP)中人群的预后差异以及预测模型对于1、3、5年预后评估的预测性能。单因素、多因素Cox回归分析用于研究TMEScore在整个人群中的预后价值,其中在单因素分析模型中具有显著差异的参数被纳入多因素分析模型中进一步验证。亚组分析用来明确TMEScore在不同的亚组人群中的预后价值。值得一提的是,该模型的预测性能也与已发表的预后模型相比较[9-11]。
1.5 列线图构建和评估 列线图是一种通过标尺的长度来衡量不同变量或者特征对结果的影响,是一种直观的风险模型可视化方法。因此,应用TMEScore整合临床变量构建预后预测的列线图用于评估1年、3年和5年的总体生存率,主要临床特征包括年龄、性别、分级、分期和TMEScore。校准曲线用于检验列线图的拟合的准确性。
1.6 富集通路分析 为了明确高、低风险评分组的富集通路差异,从MSigDB数据库中下载并获得的“c2.cp.kegg.v7.4.symbols”基因集用于GSEA分析,并比较两个风险组之间的富集通路评分差异,并用热图可视化。使用R包 “limma”“org.Hs.eg.db”“clusterProfiler”和“enrichplot”来确定高、低风险评分组之间的富集通路差异。
1.7 统计学方法 采用R(3.6.1版)和Strawberry Perl(5.32.0.1版)软件。计量资料用t检验,计数资料用卡方检验或Fisher确切概率法。预后分析采用Kaplan-Meier曲线。P<0.05为差异有统计学意义。
2 结果
2.1 鉴定HCC不同的分子亚型 在数据预处理后,370例和240例HCC样本的转录组芯片分别从TCGA和ICGC数据库中获得。通过单因素Cox分析对TCGA队列中的TME相关基因表达分析,共有528/4,061个基因被筛选为TME相关的预后基因。通过NMF分析将所有患者分成C1和C2两个亚群(见图1A),结果发现患者的OS和PFS在两个亚群中有明显的差异,C1亚群患者的OS和PFS高于C2亚群的患者(见图1B和1C)。进一步比较两个亚群之间的免疫细胞浸润评分,发现CD8+T细胞、上皮细胞、单核细胞、骨髓树突状细胞、NK细胞和T细胞在两个亚群之间有明显的差异(见图1D)。聚类分析也发现C1和C2两个亚群分别汇聚与不同的免疫亚型(见图1E)。
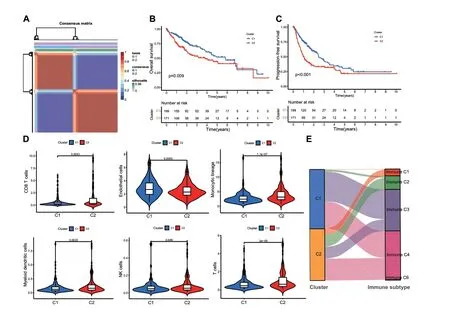
图1 两个聚类(C1和C2)生存、预后及免疫评分和亚型的比较。A. 通过非负矩阵分解算法进行聚类。B~C. 两个聚类之间的总生存期(Overall survival,OS)和无进展生存期(Progression-free survival,PFS)。D. 肿瘤微环境中的不同免疫细胞评分在两个聚类间的比较。E. 免疫亚型在两个聚类间的比例
2.2 基于TME相关基因构建预后风险评分 为了构建TME相关的预后风险评分模型,随机将TCGA队列分成训练集(224例)和测试集(146例)两组,两组间的临床基线资料差异无统计学意义(见表1)。为了验证与预后相关的TME基因,对训练集的224例患者行单因素Cox分析,共得到488个预后相关的TME基因。为了进一步缩小TME基因的范围,Lasso回归和Akaike信息准则也被用于筛选TME相关基因。最终,2个关键的TME预后基因(CDCA8和CBX2)被确认,TMEScore的风险评分模型为CDCA8的表达水平×0.3533+CBX2的表达水平×0.5156。不同TMEScore对患者的预后影响以及该评分模型对预后的诊断性能被进一步评估。单因素和多因素分析均揭示TMEScore是预后相关的风险因素(见表2)。生存分析显示基于TMEScore的高、低风险分组能够很好区分训练集(见图2A)、测试集(见图2B)、整个数据集(见图2C)以及ICGC(见图2D)中的人群预后,且高TMEScore患者比低TMEScore患者的预后更差。并且,TMEScore预测1、3和5年患者生存的AUC在训练集中为0.818、0.721和0.699(见图2E);在测试集中分别为0.671、0.652和0.633(见图2F);在整个TCGA中分别为0.762、0.704和0.664(见图2G)。在ICGC数据集中,TMEScore预测1、3和5年患者生存的AUC为0.770、0.783和0.390(见图2H)。该预测评分在ICGC数据集中的5年AUC较低的主要是由于其中位随访时间相对较短,较少的事件数影响了模型预测的效果。为了进一步分析TMEScore在不同临床特征的人群中的预测价值,亚组分析在TCGA的患者中开展。分层分析结果显示TMEScore在年龄>65岁、≤65岁、男性和女性、G1~2期、G3~4期、M0、N0、I~II期、III~IV期、T1~2期、T3~4期人群中均显示出预后预测价值(见图2I-2T)。基于TMEScore风险评分在T1期患者及其他T分期患者中差异有统计学意义,提示该预后评分对区分早期和晚期患者具有一定的价值(见图2U)。
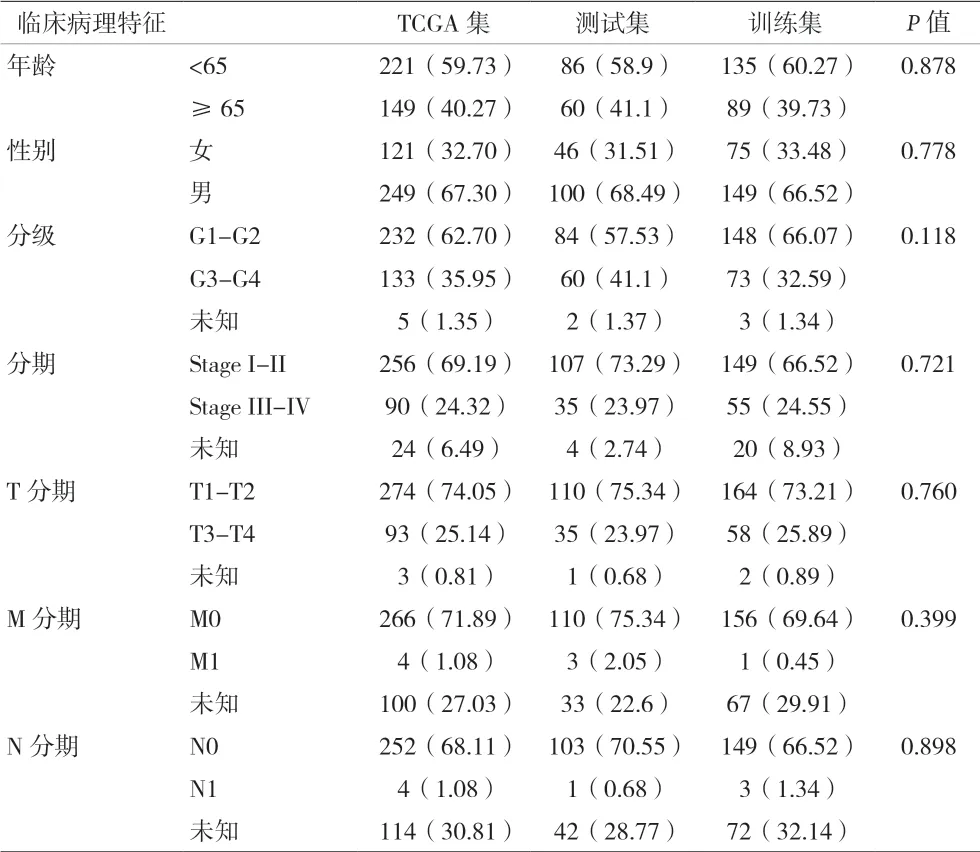
表1 训练集和测试集患者的临床基线特征[n(%)]
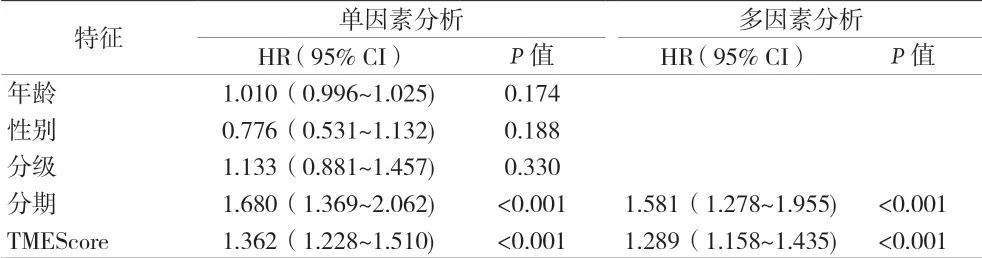
表2 单因素和多因素分析预后相关风险特征
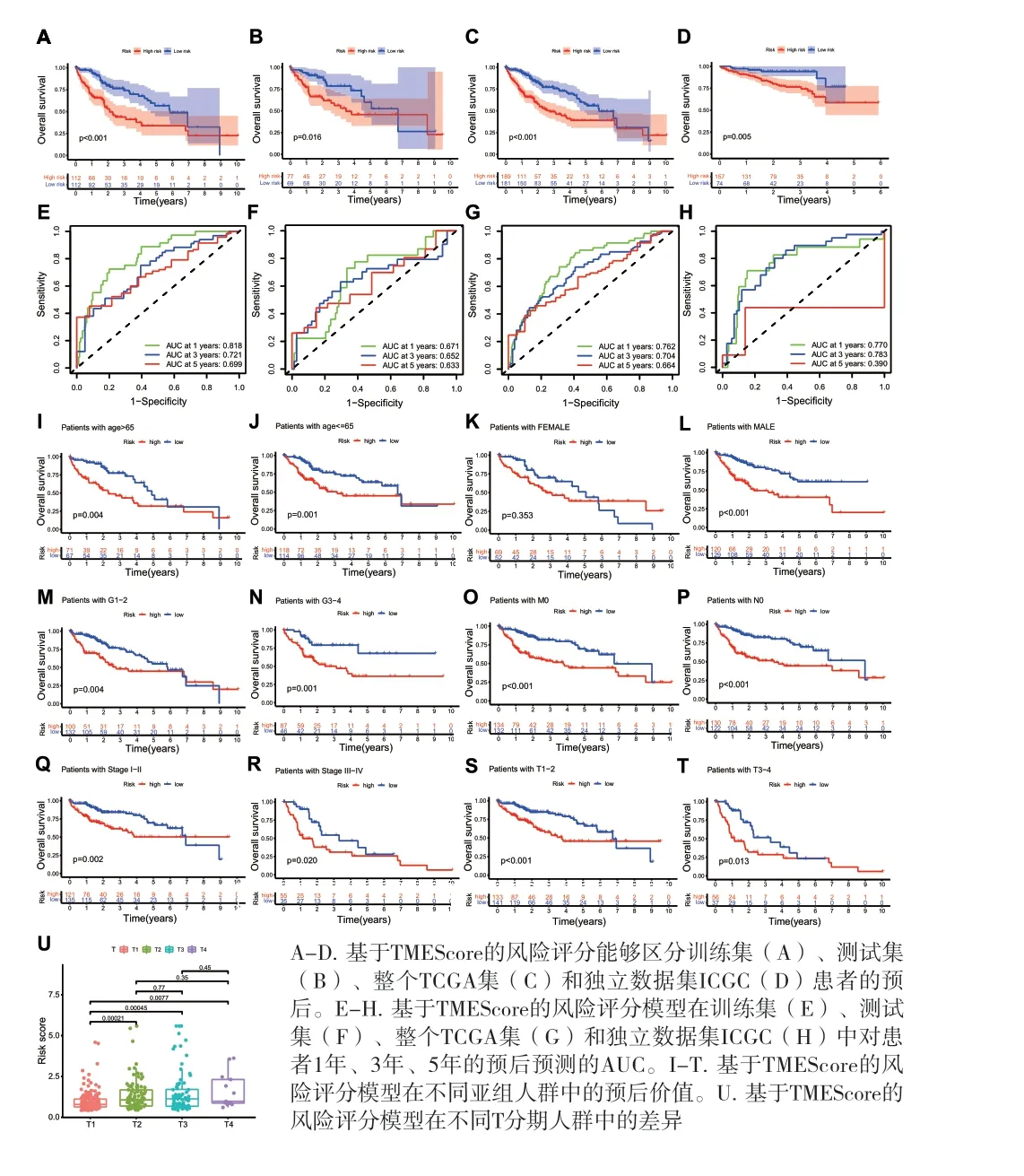
图2 基于TMEScore的风险评分的预测效能评估
2.3 整合临床参数的列线图预测预后 通过整合临床参数和TMEScore,一个预测患者1、3和5年生存评估列线图被建立(见图3A)。拟合曲线提示该列线图具有良好的拟合性(见图3B)。进一步比较构建的列线图与其他临床参数的预测性能,结果提示该列线图预测患者1年生存的AUC比其他临床参数高(AUC=0.777)(见图3C)。决策曲线分析也提示构建的列线图的预测性能比其他临床参数更好(见图3D)。为进一步验证构建的TMEScore在预测患者预后上的价值,本研究搜集了已发表的与TME相关的风险模型,并比较这些模型的预测性能。相比于其他3个已发表的TME相关预测模型,本研究的模型在1、3和5年的AUC上均高于这三个模型(见图3E)。生存分析提示Deng等、Tian等及本研究构建的模型能够良好区分高、低风险人群的预后(见图3F)。以上结果也提示本研究构建的TMEScore以及列线图模型能够有效评估患者生存预后风险,对具有高死亡风险的患者进行有效的监督随访和必要的干预对提升这些患者的生存具有重要意义。
2.4 富集通路分析和相关性分析 进一步对高、低风险评分组内的样本行基因富集分析以明确高、低风险评分组之间涉及的信号通路差异,结果提示高风险评分组与KEGG_CELL_CYCLE,KEGG_DNA_REPLICATION,KEGG_HOMOLOGOUS_RECOMBINATION,KEGG_OOCYTE_MEIOSIS和KEGG_PRIMARY_IMMUNODEFICIENCY通路相关(见图4A)。低风险评分组与KEGG_COMPLEMENT_AND_COAGULATION_CASCADES,KEGG_DRUG_METABOLISM_CYTOCHROME_P450,KEGG_FATTY_ACID_METABOLISM,KEGG_PPAR_SIGNALING_PATHWAY和KEGG_RETINOL_METABOLISM通路相关(见图4B)。进一步研究发现TMEScore与免疫检查点(PDCD1、CD274、CTLA4)、DNA复 制(POLE2、FEN1、MCM6、POLD3)、错配修复(MSH6、MSH2)和上皮-间质转化(FAP、LOXL2)均呈正相关(见图4C)。由于TMEScore与免疫检查点成正相关,进一步分析其与免疫细胞之间的相关性。结果显示TMEScore与T细胞、CD8+T细胞、细胞毒性淋巴细胞、单核细胞、髓系树突状细胞、纤维母细胞呈正相关,与中性粒细胞呈负相关(见图4D)。

图3 列线图构建可视化风险评分模型。A. 构建整合TMEScore的风险值及临床参数的列线图。B. 患者1年、3年、5年的拟合曲线。C. 不同临床参数之间预测患者1年生存的AUC比较。D. 决策曲线评估列线图的临床获益。E. 基于TMEScore的风险评分模型预测性能与先前发表的TME相关预测模型的比较。F. 基于TMEScore的风险评分模型与先前已发表的TME相关预测模型对患者的预后评估比较
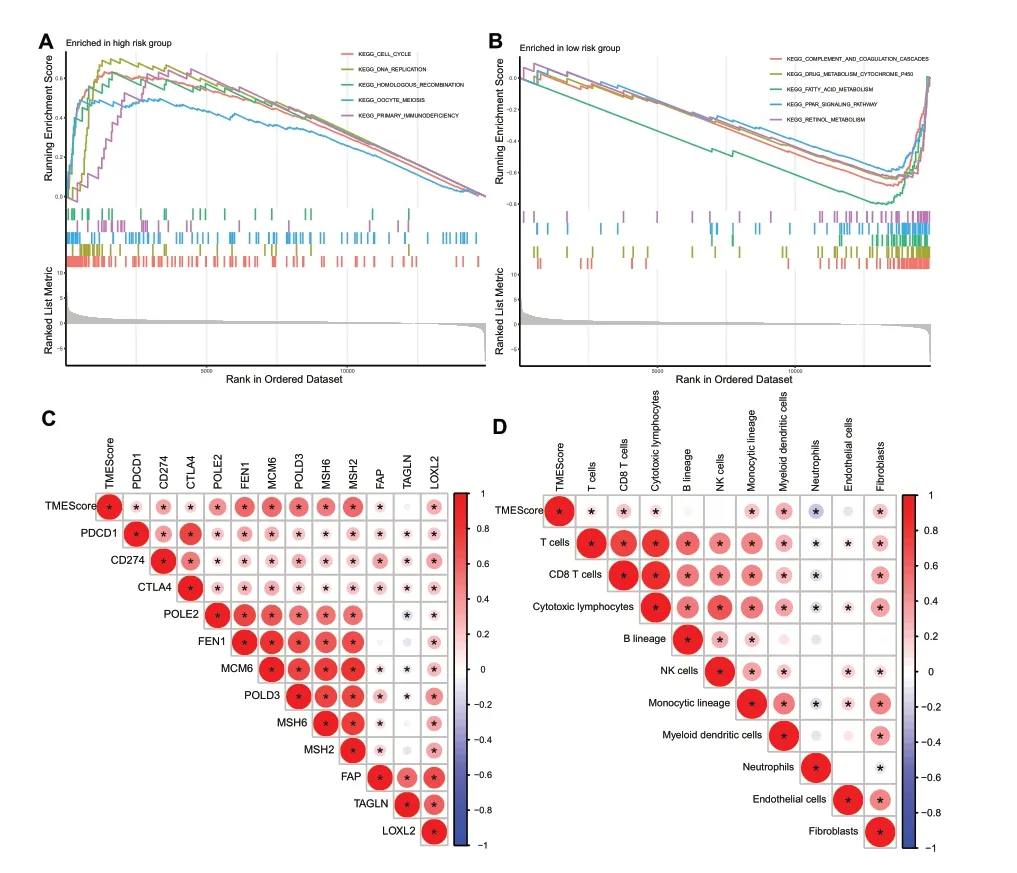
图4 基于TMEScore的风险评分值在样本中的分子机制。A-B. 高、低风险评分组中的样本参与的重要通路。C. HCC样本风险评分值与肿瘤学重要通路的靶点的代表性基因表达之间的相关性。D. HCC样本风险评分值与免疫细胞评分之间的相关性
3 讨论
大量文献表明TME在HCC的病理生成及复发、转移中起关键作用,这表明HCC的生物学行为不仅与肿瘤细胞有关,也与TME密切相关[22-23]。因此,为了明确HCC中与TME相关的预后生物标志物,本研究根据先前发表的研究整理和过滤了4,061个TME相关的基因。通过Lasso Cox回归结果确定了2个关键的TME预后基因(CDCA8和CBX2)。其中,CDCA8是染色体乘客复合体(Chromosomal passenger complex,CPC)的一个重要组成部分,与有丝分裂过程中的细胞动态定位调节有关。JOEN等[24]报道,沉默CDCA8能够恢复ATF3肿瘤抑制因子的功能和以及失活致癌信号通路AKT/β-catenin,从而抑制HCC的生长和干性,提示靶向CDCA8可能是治疗原发性HCC复发、转移的潜在靶点。CUI等[25]发现高表达的CDCA8与HCC患者更差的总生存率(P=0.0054)和无进展生存(P=0.0009)相关。体外实验显示抑制CDCA8能减缓细胞增殖,阻断G0/G1阶段的细胞周期。体内实验表明抑制CDCA8能抑制肿瘤的生长。这些发现也提示CDCA8可能是HCC的一个有效治疗靶点。另一个关键基因CBX2,是核心蛋白复合物1(polycomb repressive complex 1,PRC1)的一个重要组成部分。MAO等[26]发现高表达的CBX2与HCC患者的不良预后有关,且敲除CBX2能抑制HCC细胞的增值和促进HCC细胞的凋亡,提示CBX2是一个可能的治疗靶标。
基于这两个关键的预测子,本研究构建TMEScore并发现高TMEScore与患者更差的预后有关,且在独立数据集和亚组人群中也验证了其预后价值,提示该风险评分特征能作为预测患者预后的独立因素。基于TMEScore构建的列线图能很好评估患者的预后风险。尽管先前研究也报道TME相关基因在预后预测上的价值[9-11],但通过比较后发现本研究的模型在预测性能上高于已发表的模型。这些结果也验证了本研究的风险模型在患者预后预测上的价值。
TMEScore与免疫检查点(PDCD1、CD274、CTLA4)、DNA复制(POLE2、FEN1、MCM6、POLD3)、错配修复(MSH6、MSH2)和上皮-间质转化(FAP、LOXL2)均呈正相关。这与GSEA富集分析的结果相一致。这些重要的机制也已经被报道与HCC的发生、发展及疗效有关[27]。免疫相关分析也揭示TMEScore与多种免疫细胞相关,提示该风险评分可能与肿瘤浸润免疫细胞(tumor-infiltrating lymphocytes,TILs)水平相关。TILs水平也被认为是重要的免疫治疗生物标志物,能够反映免疫治疗疗效[28]。因此,本研究构建的风险评分模型可能对预测免疫治疗疗效也有一定的价值,但仍需要后续的研究。
本研究通过生物信息学的方法构建和验证了一个TME相关的双基因预后风险评估模型(CDCA8和CBX2)。该模型能够有效预测HCC患者预后风险水平,对于具有高死亡风险的人群,临床医师能够采取必要的监督随访和合理的干预措施提升患者的生存。
