Understanding the Pathogenesis of Generalized Pustular Psoriasis Based on Molecular Genetics and lmmunopathology
2023-01-12AnQiZhaoandMingLi
An-Qi Zhao and Ming Li,3,*
1 Department of Dermatology, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200092, China;2 Institute of Dermatology, School of Medicine, Shanghai Jiao Tong University, Shanghai 200092, China; 3 Department of Dermatology, The Children’s Hospital of Fudan University, Shanghai 200092, China.
Abstract Generalized pustular psoriasis (GPP) is a rare and life-threatening autoinflammatory skin disease characterized by recurrent and sudden episodes of widespread rashes with scattered sterile pustules. Clinical and genetic evidence indicates that the pathogenesis of GPP both overlaps and is separate from psoriasis vulgaris (PV). Interleukin (IL)-23/IL-17 immune pathway is well known to play a critical role in the immunopathogenesis of PV, while the inflammation of GPP is more inclined to involve the innate immune response via the IL-1/IL-36–chemokine pathway. Mutations in IL36RN, CARD13, AP1S3, MPO, TNIP1, SERPINA3, and SERPINA1 have been shown to be associated with GPP,among which loss-of-function mutation in IL36RN is the dominant mutation with the highest prevalence. Recent studies have shown that interaction of the IL-36 pathway and the IL-23/IL-17 axis underlies the immunological disturbances of GPP, indicating that innate and adaptive immune responses intertwine in the pathogenesis of GPP.With this deeper understanding of the pathogenesis of GPP, treatment by biologics targeting the IL-1/IL-36 pathway appears to be promising. IL-1 inhibitors, anakinra, canakinumab, and gevokizumab have reportedly been effective in some cases. Spesolimab and imsidolimab, which are antibodies to the IL-36 receptor, are undergoing investigation in a phase II trial and showing promising results. In the present review, we illustrate the current understanding of the pathogenesis of GPP based on recent updates on the molecular genetics and immunopathology of GPP and review recent clinical trials and case reports of novel biologics in the treatment of GPP.
Keywords: autoinflammation, generalized pustular psoriasis, GPP, genetic mechanisms, IL-36
lntroduction
As a group of inflammatory skin diseases characterized by the development of neutrophilic pustules, pustular psoriasis (PP) was historically grouped with psoriasis vulgaris (PV) because of their concurrence. However,PP is now considered a separate entity as research into its pathogenesis has intensified. PP is divided into 3 subgroups based on the lesion scope: generalized PP (GPP)(also known as von Zumbusch disease), palmoplantar pustulosis (PPP), and acrodermatitis continua of Hallopeau (ACH).1In 2017, the consensus statement on phenotypes of PP formulated by the European Rare and Severe Psoriasis Expert Network (ERASPEN)-labeled primary pustular conditions as PP and used the term “psoriasis with pustules” to differentiate PP from pustules within or at the edge of plaque psoriasis.2
GPP was first described by von Zumbusch in 1920, but a consistent definition has not been established. GPP is a potentially life-threatening autoinflammatory disease characterized by periodic flares of widespread erythema with visible primary sterile pustules on nonacral skin.It can develop at any age but mostly occurs in the fifth decade of life, with a mean age of 45.6 to 50.0 years at diagnosis. Affected patients show a female preponderance.3GPP can occur with or without systemic inflammation and/or PV.1,2-4According to the ERASPEN consensus,GPP should only be diagnosed when the condition has recurred at least once or lasts for >3 months, and acute generalized exanthematous pustulosis should be actively excluded.2
The pathogenesis of GPP is not completely understood,but it is expected to involve genetic and environmental factors similar to PV and other inflammatory skin diseases. Provocative factors of a flare mainly include medications, especially the withdrawal of corticosteroids or cyclosporine; various infections; pregnancy; and electrolyte imbalance.1The identification ofIL-36RNmutations in patients with GPP has highlighted the pathogenic potential of the interleukin (IL)-1/IL-36 axis.5Various findings have indicated that the autoinflammation driven by the IL-1/IL-36–chemokine–neutrophil axis plays a crucial role in the pathogenesis of GPP.6-7Innate inflammation cooperates with the adaptive immune response induced by the tumor necrosis factor (TNF)-α/IL-23/IL-17/IL-22 axis, leading to a progressive cycle of inflammation. In 2019, Akiyama8proposed that early-onset GPP due toIL36RNmutations can be classified as an autoinflammatory keratinization disease (AiKD). However, patients withIL36RNmutations account for a minority of patients with GPP, with studies showing a frequency of 8.3%among 84 patients with GPP9and 23.7% among 190 patients with GPP.10This indicates that other mutations may affect IL-1/IL-36 signaling. For these patients with GPP who lackIL-36RNmutations, the immune mechanisms driving disease onset are unclear. In the last decade,more gene mutations includingCARD14,AP1S3,MPO,TNIP1,SERPINA3, andSERPINA1have been successively detected in patients with GPP, enabling us to further explore its pathogenesis. Patients harboring disease alleles at 2 distinct loci (IL36RNandAP1S3;IL36RNandCARD14) have also been reported.11-12Mössneret al.11reported a 15-times higher frequency of heterozygous carriers ofIL36RNmutations in patients with GPP(8%) than in the general population (0.54%). Among 15% of these carriers,IL36RNmutations occurred with additional variants in other genes. These identifications indicate complex inheritance of GPP that is probably oligogenic rather than monogenic.
With a focus on the molecular genetic mechanisms of GPP, PubMed was searched from 2016 to 2021 for relevant articles published in the English language using a combination of the following keywords: ([generalized pustular psoriasis] or [GPP]) and ([pathogenesis] or[mechanism]). Three researchers screened the titles and abstracts and then reviewed the full text for inclusion.The present review aims to illustrate the pathogenesis of GPP, which involves autoinflammatory responses and adaptive immune responses, and to introduce novel biological agents targeting the critical IL-1/IL-36 pathway in the development of GPP.
Mutations causing autoinflammatory responses in GPP
Gene mutations associated with GPP have been identified inIL36RN,CARD14,AP1S3,MPO,TNIP1,SERPINA3,andSERPINA1. These mutations have been discovered to be associated with the IL-1/IL-36 pathway.IL36RN,CARD14, andAP1S3gene mutations activate proinflammatory signaling pathways via nuclear factor-κB(NF-κB), which further results in increased expressions of chemokines and IL-36 proinflammatory cytokines.MPO,SERPINA3, andSERPINA1loss-of-function (LOF)mutations increase protease activity and in turn activate proinflammatory IL-36 pathways. In gene expression analysis of lesional biopsies, GPP lesions had higher levels of IL-36 inflammatory cytokines than did PV lesions.7This evidence suggests that the autoinflammation via the IL-36 signaling pathway may be pivotal in the pathogenesis of GPP. The inflammatory responses induced by gene mutations are shown in Fig. 1.
GPP and IL36RN LOF mutations
Molecular mechanisms underlying IL36RN mutations
IL36RNencodes IL-36 receptor antagonist (IL-36Ra).As members of the IL-1 family, IL-36 cytokines (comprising 3 agonists [IL-36α, IL-36β, and IL-36γ] and 2 antagonists [IL-36Ra and IL-38]) have the same receptor (ie,the IL-36 receptor [IL-36R]). They act as regulators of the innate immune system and are expressed in epithelial and immune cells, especially in keratinocytes. The IL-36 pathway is activated after the binding of IL-36 agonist to IL-36R, leading to the activation of the intracellular NF-κB and mitogen-activated protein kinase pathways. In vitro functional assays have shown thatIL36RNpathogenic mutations lead to decreased expression or activity of IL-36Ra, which results in the inability of IL-36Ra to antagonize and limits the proinflammatory effects.13The unbalanced IL-36 pathway activates the NF-κB and mitogen-activated protein kinase pathways. This further leads to increased expression of proinflammatory cytokines including inflammatory cytokines (IL-6, IL-1β, IL-23,and TNF-α), neutrophil chemokines (CXCL1, CXCL2,CXCL6, and CXCL8), and lymphokines (CCL20 and CCL3), as well as the proliferation of T cells and dendritic cells, contributing to the neutrophilic infiltration in skin pustules and systemic inflammation in patients with GPP.13
Identification of IL36RN mutations in patients with GPP
In 2011, Marrakchiet al.5first identified the c.80T>C(p.Leu27Pro) homozygous missense mutation in theIL36RNgene in 9 Tunisian families that were segregated with autosomal recessive GPP. They also demonstrated that the abnormal IL-36Ra structure and function caused by p. Leu27Pro mutations led to uncontrolled secretion of inflammatory cytokines in GPP.5A series ofIL36RNvariants were subsequently identified in patients with GPP. The main variant types were missense mutations and nonsense mutations, as well as 1 splicing mutation (c.115 + 6T>C)and 2 small deletion mutations (c.420_426del and c.295-300del). c.115 + 6T>C is the most common mutation in Asian populations, whereas c.338C>T is the most prevalent in Europeans.9Liet al.14demonstrated that the c.115 + 6C>T mutation rate in the normal population was 3.6% (13 of 365 cases), with 2 homozygous mutations in 13 cases. This finding indicates that multiple factors might contribute to the pathogenesis of GPP.
Genotype–phenotype characteristics of IL36RN mutations

Figure 1. Inflammatory responses induced by gene mutations. The innate immune response via the IL-1/IL-36-chemokine pathway plays the central role in the pathogenesis of GPP (shown by the bolded arrow). IL-36RN loss-of-function mutations lead to the deficiency of IL-36 receptor antagonists (IL-36Ra), inducing the more active binding of the IL-36 receptor to IL-36 agonists. MPO and SERPINA 1/3 loss-offunction mutations lead to the activation of proteinase 3, cathepsin G/S, and elastase; these proteases participate in the processing of IL-36 precursors, turning them into active IL-36 cytokines. Thus, IL-36RN, MPO, and SERPINA 1/3 mutations lead to the upregulation of IL-36 signaling, which further activates the downstream proinflammatory NF-κB and MAPK pathways. AP1S3 and TNIP1 loss-of-function mutations and CARD14 gain-of-function mutations are involved in IL-36 signaling by hyperactivating the NF-κB pathway. The inflammatory responses induce IL-36 expression in an autocrine manner and promote the recruitment of neutrophils, T cells, and monocytes by the production of proinflammatory cytokines (IL-1β, IL-23, IL-6, and TNF-α), neutrophil chemokines (CXCL1, CXCL2, and CXCL8), lymphokines (CXCL3 and CXCL20), and co-stimulatory molecules by dendritic cells and T cells. Interestingly, the upregulated IL-36 signaling in GPP promotes the proliferation of IL-17-producing CD4+Th17 cells, and these cytokines secreted by infiltrating CD4+T cells further propagate these inflammatory cycles by inducing expression of IL-36 and other inflammatory mediators. GOFM: gain-of-function mutation, GPP: generalized pustular psoriasis, IL: interleukin, LOFM: loss-of-function mutation, MAPK: mitogen-activated protein kinase, NF-κB: nuclear factor-κB, TNF: tumor necrosis factor.
GPP can be divided into GPP alone and GPP with PV.GPP alone appears to be an independent entity of psoriasis that differs from PV because of its higher rate ofIL36RNmutation. In previous studies of the Chinese Han population, 46.8% to 60.5% of patients with GPP showedIL36RNmutation, and the mutation rate in patients with GPP without PV was significantly higher(76.6%–79.2%) than that of patients with both GPP and PV (36.8%–37.8%).15-16Studies have also shown high mutation rates ofIL36RNin patients with GPP without PV in Japan (81.8%)17and Germany (46.2%).18
Liet al.14first assessed the mutation rates ofIL36RNin patients with pediatric-onset GPP (PGPP) and adult-onset GPP (AGPP) and found that PGPP had a stronger hereditary susceptibility (66.7% in PGPPvs. 34.2% in AGPP,P= 0.008). c.115 + 6T> C was the most common mutation and showed a significant difference in the prevalence between the PGPP and AGPP groups (66.7%vs.28.9%,P= 0.0035).14Wanget al.19assessed the different clinical profiles of PGPP withIL36RNmutations and demonstrated that the variation c.115 + 6T>C was the most common mutation in PGPP (63.6%), followed by the p.Pro76Leu mutation (10.6%). Lianget al.20demonstrated that the c.115 + 6T>C variant was significantly associated with the presence of geographic tongue in patients with GPP who hadIL36RNmutations.
Hussainet al.21compared the clinical features of patients with GPP who had recessiveIL36RNalleles(n= 49) and those who had no pathogenic alleles at this locus (n= 166). The authors found thatIL36RN-positive patients presented a more severe clinical phenotype characterized by an earlier age at onset (17 ± 2.4vs. 33 ± 1.5 years,P= 5.9 × 10-7), a higher risk of systemic inflammation (83.3%vs. 55.6%,P= 1.5 × 10-3), and a lower prevalence of PV (36.1%vs. 68.7%,P= 5.0 × 10-4).21A severe clinical phenotype ofIL36RN-positive cases was also demonstrated in an ambispective study of 66 patients with PGPP conducted by Wanget al.19In 2017,they proposed 3 minor signs suggestive ofIL36RNmutations: confluent lakes of pus (P= 0.002), perianal erosion (P= 0.014), and flexural erosion (P= 0.007). More patients with than without theIL36RNmutation converted to ACH (P= 0.029). In addition, the age at onset was nearly halved in the homozygotes/compound heterozygotes compared with theIL36RN-negative patients.19In 2018, Mössneret al.11reported that the age at onset was significantly lower inIL36RNmutation carriers than in noncarriers (P= 6.9 × 10-3), especially in biallelic mutation carriers (P= 7.4 × 10-4). More recently, the dose-dependent effect ofIL36RNdisease alleles on age at onset in all forms of PP (P= 0.003) was proposed by Twelveset al.10
GPP and CARD14 gain-of-function mutations
CARD14encodes caspase recruitment domain family member 14 (CARD14, also known as CARMA2), which mediates TRAF2-dependent activation of NF-κB signaling. As a scaffold protein, it is specifically expressed in the skin and predominantly localized in keratinocytes.Gain-of-functionCARD14variants lead to upregulation of NF-κB signaling, inducing inflammatory responses.22In 2012, Jordanet al.23identified the de novoCARD14gain-of-function variant c.423A>C (p.Glu138Ala) in a European child with severe GPP. CARD14 expression is upregulated in the granular layers of GPP-affected skin,whereas it is mostly restricted to the basal layer of the healthy skin epidermis.23TheCARD14c.526G>C (p.Arg179His) heterozygous missense mutation was identified as a significant risk factor for GPP in patients with PV in a Japanese cohort.24Qinet al.25first analyzed the variants ofCARD14in a Chinese Han population with PV and GPP. They identified c.355A>G (p.Met119Val), c.497G>A(p.Arg166His), and c.2044C>T (p.Arg682Trp) in patients with concurrent GPP and PV and proposed a significant association ofCARD14variants with GPP.25Liet al.26investigated theCARD14variants in GPP with PV (121 patients) and GPP alone (48 patients) and reported a differentCARD14genetic background with variant carrier rates of 9.9% in the GPP with PV group versus 0% in the GPP alone group (P= 0.020). The authors suggested thatCARD14variants may play an essential role in the pathogenesis of PV-related disease.26Further correlations betweenCARD14mutations and the onset of GPP remain to be studied.
GPP and AP1S3 mutations
AP1S3encodes the σ-1C subunit of the activator protein 1 (AP-1) complex, a conserved heterotetramer that promotes vesicular trafficking between the trans-Golgi network and the endosome. The σ1 subunit confers stability to AP-1 tetramers; thus,AP1S3mutations are expected to disrupt the entire complex. In 2014, Setta-Kaffetziet al.27first reported 2 founder mutations of heterozygousAP1S3(c.11T>G [p.Phe4Cys] and c.97C>T [p.Arg33Trp]) in European patients with PP, including GPP, ACH, and PPP, but not in Asian or African patients with GPP.Twelveset al.10demonstrated anAP1S3-variant prevalence of 10.8% (4/37) in European patients with GPP.Setta-Kaffetziet al.27proposed that the variants destabilized the structure of the AP-1 complex, thus disrupting the endosomal translocation of the innate pattern-recognition receptor Toll-like receptor 3 and inhibiting the downstream signaling. Then, in 2016, Mahilet al.12proposed thatAP1S3mutations disrupted keratinocyte autophagy, causing an abnormal accumulation of p62 and the activation of NF-κB signaling. This alteration results in upregulation of IL-1 signaling and overexpression of IL-36α (among other cytokines), which is an important mediator of skin inflammation.12
GPP and MPO LOF mutations
TheMPOgene encodes the neutrophilic enzyme myeloperoxidase (MPO), a lysosomal hemoprotein located in the azurophilic granules of polymorphonuclear leukocytes and monocytes. MPO is endowed with potent antimicrobial oxidizing capabilities once activated. In 2020,Haskampet al.28demonstrated thatMPOgene defects play a role in the pathogenesis of GPP through regulating protease activity and neutrophil extracellular trap formation and modifying efferocytosis, thus activating proinflammatory IL-36 signaling as a result.
AlthoughMPOdeficiency was reported in single persons with GPP decades ago, it was not considered as a disease gene of GPP until 2020, when Vergnanoet al.29identified the homozygous splicing mutation c.2031-2A>C inMPOafter whole-exome sequencing of 19 unrelated individuals with GPP. They also reportedMPOmutations (c.2031-2A>C and c.1705C>T compound heterozygous mutations and c.1552_1565del homozygous mutation) in phenotypically GPP-related diseases and further demonstrated that c.2031-2A>C and c.1705C>T disease alleles were associated with increased neutrophil abundance in the general population.29Subsequently,Haskampet al.28identified 8MPOvariants in 15 patients with GPP, including 3 homozygous variant carriers (c.265_275dup11, c.2031-2A>C, and c.1768C>T),1 compound heterozygous variant carrier (c.995C>T and c.2031-2A>C), and 11 heterozygous variant carriers (c.752T>C, c.995C>T, c.2031-2A>C, c.1705C>T,c.1642C>T, and c.1555_1568del).
Studies also showed that affected individuals with homozygous or compound heterozygousMPOmutations had a lower onset age than those with bothIL36RNandMPOmutations (P= 0.0018), implying that the number ofMPOmutant alleles correlates with age at onset.27-28
GPP and TNIP1 mutations
TheTNIP1gene encodes TNFAIP3-interacting protein 1 (TNIP1). TNFAIP3 is TNF-α induced protein 3,which works downstream of TNF-α to regulate NF-κB signaling. The association ofTNFAIP3andTNIP1gene polymorphisms with psoriasis susceptibility has been confirmed in people of multiple ethnicities.30To further investigateTNIP1gene polymorphisms and GPP, Hanet al.31focused on the Chinese Han population and reported that the frequencies of alleles of 5 SNPs were significantly different between the patients who had GPP without PV and the control group. They suggested thatTNIP1might be a susceptibility gene for GPP.31TNIP1andTNFAIP3are also susceptibility genes for multiple autoimmune diseases, including rheumatoid arthritis, ulcerative colitis,Crohn disease, psoriasis, and systemic lupus erythematosus, suggesting their roles in the pathogenesis of autoimmune disorders.
GPP and SERPINA3, SERPINA1 LOF mutations
SERPINA3encodes serine protease inhibitor A3 (serpin A3), which inhibits several proteases including the neutrophil serine protease CTSG, an enzyme that activates IL-36β precursors to the more active (>500 times) IL-36β cytokine. The impaired function of serpin A3 probably results in less inhibition of CTSG and more active IL-36 cytokines, contributing to the IL-36-driven inflammation in GPP.32In 2020, Freyet al.32first detected a heterozygous deletion c.966delT (p.Tyr322Ter) in 2 of 25 patients with GPP that caused an LOF variant inSERPINA3, and the authors proposedSERPINA3as a new candidate gene for GPP. Soon after, Kantaputraet al.33reported 2 unrelated patients, both of whom carried the same heterozygous variant c.718G>A (p.Val240Met) inSERPINA1,one with adult-onset immunodeficiency syndrome and the other with GPP. SERPINA1 is a major plasma serine protease inhibitor and shares approximately 40% to 45% of amino acids and inhibitory functions with SERPINA3,33indicating that SERPINA family members may be implicated in the pathogenesis of GPP.
Cooperation of innate and adaptive immune responses in the pathogenesis of GPP
The pathogenesis of psoriasis is complex and unclear,and it involves both the innate and adaptive immune systems. Antimicrobial peptides including cathelicidin, human β-defensins, S100 protein, lipocalin 2, and RNase 7 are an integral part of the first-line defense of a host against pathogens, and they have been illustrated to be important immunomodulators in the progression of psoriasis.34With their high expression in psoriatic skin lesions, antimicrobial peptides can broadly regulate both innate and adaptive immunity by interacting with various immune cells.35The identification ofIL36RNmutations in patients with GPP has highlighted the IL-36 family cytokines as critical mediators in the pathogenesis of GPP and has characterized GPP as an autoinflammatory condition. Disease alleles associated with GPP, includingIL36RN,CARD14,AP1S3,MPO,TNIP1,SERPINA3, andSERPINA1, have been subsequently identified and reported to be involved in IL-1/IL-36 signaling, further underscoring the critical role of autoinflammatory responses that contribute to the pathogenesis of GPP (Fig. 2).
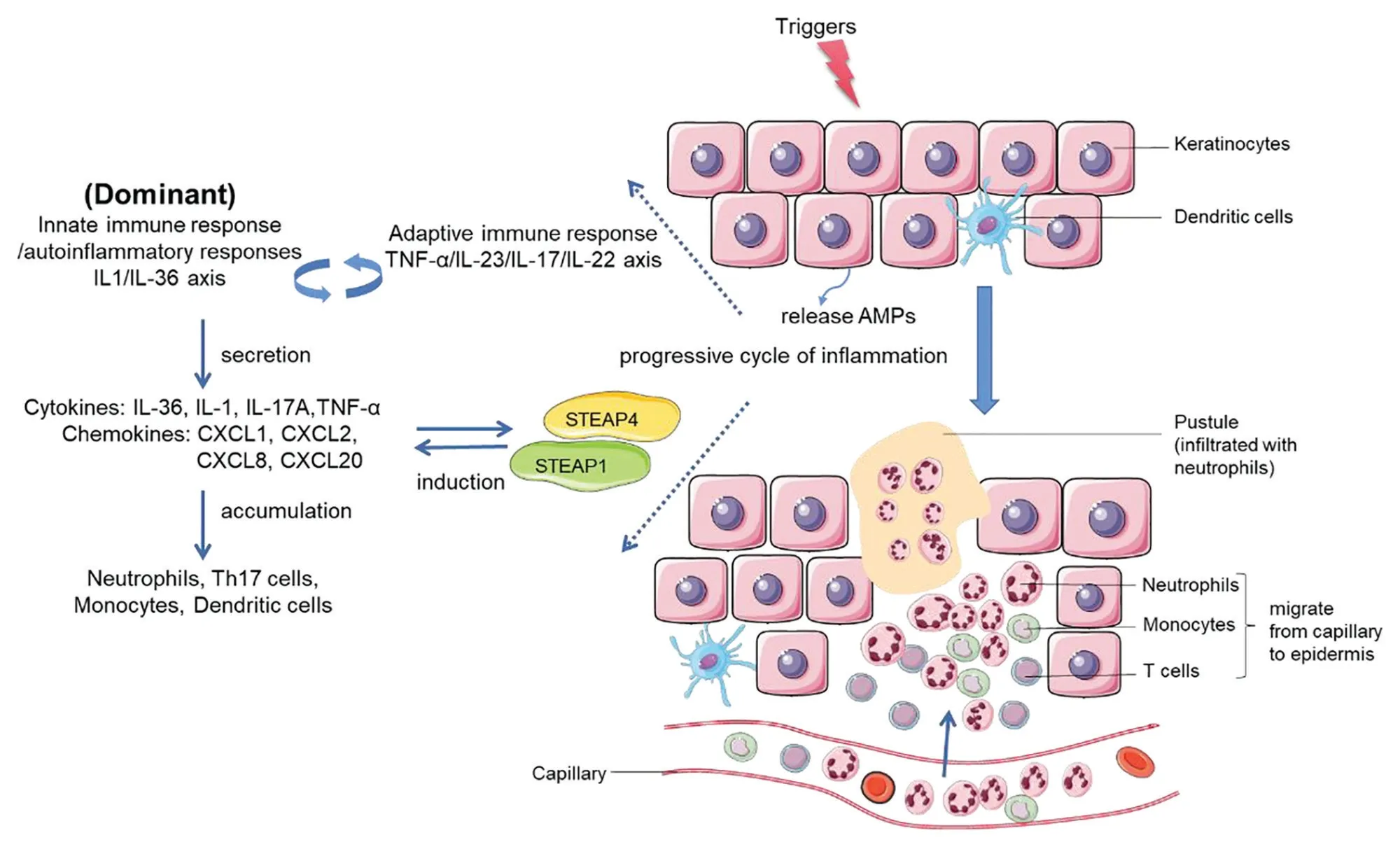
Figure 2. Progressive cycle of inflammation in the pathogenesis of GPP. Keratinocytes can be stimulated by initial triggers, and stressed keratinocytes release AMPs. AMPs in turn interact with various immune cells and are involved in innate and adaptive immune responses.The IL-1/IL-36 axis (autoinflammatory response/innate immunity), as the critical driving pathway, cooperates with the TNF-α/IL-23/IL-17/IL-22 axis (adaptive immune response). They form a positive inflammatory feedback loop and further induce the activation and secretion of chemokines/cytokines such as IL-36, IL-1, IL-17A, TNF-α, CXCL1, CXCL2, CXCL8, and CXCL20. STEAP1 and STEAP4, 2 inflammatory-related proteins, amplify the activation of proinflammatory cytokines. The above interactions ultimately form a vicious circle of enhancing inflammation, leading to the accumulation and activation of neutrophils (mainly), T cells and dendritic cells, which migrate from capillaries to the epidermis, histopathologically manifesting as the infiltration of massive neutrophils. AMPs: antimicrobial peptide, IL: interleukin, STEAP:six-transmembrane epithelial antigens of the prostate, TNF: tumor necrosis factor.
To better understand the mechanism of GPP and PV,Johnstonet al.7performed a gene expression study on GPP (n= 28) and PV (n= 12) lesional biopsies and healthy control (n= 20) skin. The authors detected significant contributions of IL-17A, TNF-α, IL-1, IL-36,and interferon-γ in both diseases.7GPP lesions showed greater expression of IL-1 and IL-36, mainly in keratinocytes surrounding neutrophilic pustules; by contrast, the expression levels of IL-17A and interferon-γ were significantly increased in PV, indicating that GPP pathways both overlap and are separate from PV.7The IL-23/IL-17 immune pathway is well known to play a critical role in the immunopathogenesis of PV. Different from the pathogenesis of PV, the inflammation of GPP is more inclined to involve the innate immune response with hyperactivation of the IL-36 axis. Whether from the higher levels of IL-1β, IL-36α, IL-36γ expression,and neutrophil chemokines (CXCL1, CXCL2, CXCL6,and CXCL8) detected in GPP lesions7or from the discovery ofIL-36RNLOF mutations and other mutations associated with the activation of IL-36 signaling as mentioned above, powerful evidence supports the notion that the IL-1/IL-36 inflammatory axis is a potent driver of GPP-related autoinflammation, and initial genetic factors related to the hyperactivation of innate immunity play important roles in the pathogenesis of GPP. The pivotal role of the IL-1/IL-36 inflammatory axis was also clarified by a gene expression study on skin lesions of patients with pustular skin diseases (GPP,n= 30; PPP,n= 17; and acute generalized exanthematous pustulosis,n= 14) performed by Lianget al.36in 2017. They found that 2 six-transmembrane epithelial antigens of the prostate (STEAP) proteins, STEAP1 and STEAP4, were upregulated in patients’ lesions and colocalized with IL-36γ around neutrophilic pustules, whereas they were not overexpressed in the lesions of patients with PV.36Further analysis showed that overexpression of STEAP1 and STEAP4 was associated with the activation of proinflammatory cytokines, including IL-1, IL-36, CXCL1,and CXCL8, indicating that they may positively regulate the induction of neutrophil chemokines and proinflammatory cytokines in PP.36
A mechanistic link of the IL-1/IL-36 axis and IL-23/IL-17 axis has been illustrated in patients with psoriasis and autoimmune disorders such as rheumatoid arthritis and Crohn disease.37Muhret al.38found that IL-17 induced IL-36α, IL-36β, and IL-36γ more potently in human psoriasis-derived keratinocytes than in healthy keratinocytes. In psoriasis-like animal models,Carrieret al.39showed that IL-17A, IL-22, and TNF-α were potent inducers of IL-36 cytokines and that IL-36α,IL-36β, and IL-36γ could upregulate the expression levels of TNF-α, IL-6, and IL-8, respectively, which were further augmented by the simultaneous addition of IL-17A or TNF-α. The heterogeneity of a genetic predisposition to GPP and the effectiveness of clinical drugs interfering with CD4+T-cell activation or the IL-23/IL-17 axis suggest that the role of T cells in the GPP pathogenesis should not be ignored.11,40-41Arakawaet al.6investigated the involvement of T cells in the GPP pathogenesis and reported that the unopposed IL-36 signaling in GPP promoted the proliferation of IL-17-producing CD4+Th17 cells, revealing the pathogenic pathway in which innate immune dysregulation promotes T-cell-mediated inflammation in GPP.
Novel biologics for GPP as an AiKD
The establishment of standard guidelines for GPP treatments remains challenging because of the overall rarity of this disease. Treatment should be based on the extent of involvement and severity of the disease.42Biologics targeting cytokines of the TNF-α/IL-23/IL-17 axis have been used to treat GPP according to clinicians’ experience in treating PV. The treatment response in patients with GPP is heterogeneous. Recently, based on novel cognition of the pathogenesis of GPP, inhibitors of the IL-1/IL-36 axis have been studied as novel promising therapeutic targets.
IL-1 inhibitors, including the recombinant IL-1α receptor antagonist anakinra,43as well as the IL-1β monoclonal antibodies canakinumab44and gevokizumab,45have shown efficacy in treating GPP. In one report, a patient carrying a c.142C>T and c.338C>T compound heterozygousIL36RNmutation exhibited lesion clearance 6 weeks after receiving 9 days of subcutaneous anakinra at 100 mg daily.43Gevokizumab, a monoclonal antibody to IL-1β, was reportedly effective in the treatment of 2 patients with severe GPP, with a respective 79% and 65% reduction in the GPP lesion area and severity index scores.45Skendroset al.44described a patient with severe GPP who discontinued anakinra because of persistent hypersensitivity skin reactions and finally achieved a complete and sustained response to the anti-IL-1β monoclonal antibody canakinumab. Large prospective randomized clinical trials in patients with GPP are needed to confirm the broad efficacy and safety of IL-1-targeted therapies for the treatment of GPP flares.
Blockade of IL-36R signaling is another appealing targeted therapeutic approach for patients with GPP.Spesolimab (BI 655130) is a selective, humanized antibody against the IL-36 receptor. A phase I proof-of-concept, open-label study led by Bachelezet al.46showed that after receiving an intravenous dose of 10 mg/kg of spesolimab, 5 of 7 patients presenting with a GPP flare had rapid (within 7 days) and sustained (up to 20 weeks)improvements in their clinical signs and symptoms as assessed by the GPP Physician Global Assessment(GPPGA) with a score of 0 or 1 as an endpoint. Among these 7 patients, 2 had a homozygousIL36RNmutation, 1 had a homozygousIL36RNmutation and a heterozygousCARD14mutation, and the remaining 4 did not show mutations inIL36RN,CARD14, orAP1S3.Spesolimab was effective in these patients, indicating that the IL-36 pathway is critical to the pathogenesis of GPP regardless of a patient’s genetic background. Bachelezet al.47further conducted a phase II randomized trial involving 53 patients with GPP receiving a single 900-mg intravenous dose of spesolimab (n= 35) or placebo(n= 18). The results showed that the spesolimab group had a higher incidence of lesion clearance at 1 week with a GPPGA pustulation subscore of 0 than did the placebo(54.3%vs. 5.6%,P< 0.001); however, spesolimab was associated with infections and systemic drug reactions.47A phase II, multicenter, double-blind, randomized, placebo-controlled trial of spesolimab in patients with GPP is ongoing, which will further evaluate the efficacy,safety, and tolerability of spesolimab.48Other anti-IL-36 therapies are also under development. Imsidolimab(ANB019), a monoclonal antibody targeted against the IL-36 receptor, achieved positive results in a single-arm,open-label, phase II trial of 8 patients with GPP. Six of them (75%) achieved the primary endpoint of response on the Clinical Global Impression scale at weeks 4 and 16. Genotypic testing indicated homozygous wild-typeIL-36RN,CARD14, andAP1S3alleles in all 7 tested patients.49A phase III trial called GEMINI-1 has also been initiated, which will enroll 45 patients with GPP and divide them into 3 groups to receive a single dose of 750 mg intravenous imsidolimab, 300 mg intravenous imsidolimab, or placebo.50GEMINI-2 will be subsequently carried out with re-enrollment of the patients from GEMINI-1 based on their response to imsidolimab,aiming to assess the efficacy and safety of imsidolimab after 6 months of monthly dosing (200 mg of subcutaneous imsidolimab).50
Conclusion and limitations
Since the discovery of some pathogenic gene mutations in patients with GPP (first theIL-36RNmutation followed by others [CARD14,MPO, andAP1S3]), great progress has been made in elucidating the genetic molecular mechanism. Research has emphasized the important role of initial genetic factors related to autoinflammation via the IL-1/IL-36 axis, defining GPP as a representative AiKD. In addition, GPP skin lesions are characterized by abnormal infiltration of neutrophils and high expression of IL-17,which proves that the TNF-α/IL-23/IL-17/IL-22 axis also participates in its pathogenesis. Arakawaet al.6proposed a close interaction between the IL-36 pathway and Th17 responses in patients with GPP, which was further confirmed by the treatment response to biological agents. In conclusion, innate immune and adaptive immune pathways are closely related and mutually reinforced in the pathogenesis of GPP.
With this deeper cognition of the pathogenesis of GPP, progress in the treatment of GPP with biologics has also been encouraging. New biologics targeting the IL-1/IL-36 axis appear promising. Case reports and clinical trials suggest that these biologics can effectively control skin inflammation and improve the quality of life of some patients with GPP. However, the treatment response in patients with GPP is heterogeneous.Relevant studies are limited to trials with small samples and short-term follow-up; thus, clinical studies with larger sample sizes and multiple ethnicities are necessary to obtain reliable data on the efficacy and longterm safety of biologics.
This review has 2 main limitations. First, it was based on the authors’ own analysis and summary of the literature and is subjective in nature. Second, this review only covered research published in mainstream journals indexed in PubMed during the last decade.
杂志排行

国际皮肤性病学杂志的其它文章
- Efficacy and Safety of lxekizumab in Chinese Patients With Moderate-to-Severe Plaque Psoriasis: 60-Week Results From a Phase 3 Study
- Characterization of Generalized Pustular Psoriasis in Northwest China: A Single-Center Retrospective Study
- Perspective on Melanoma in the Arab World:A Quantitative and Qualitative PubMed-Based Analysis of Research Output (2004–2019)
- Laboratory Safety of Dupilumab, and lts Effect on lnflammatory Biomarkers, in Chinese Adults With Moderate-to-Severe Atopic Dermatitis: An Analysis of a Randomized, Double-Blind Phase lll Study
- Sexual Behavior and Awareness of Sexually Transmitted Diseases Among Street-Based Female Sex Workers in the Florence Area, Central Italy
- Perceptions of Acne and Its Treatments Among Chinese College Students: A Cross-Sectional Survey