甲醛在本征和金掺杂石墨烯表面的吸附:第一性原理研究
2022-12-26刘佳鑫张仟春
唐 珊,姜 丽,,刘佳鑫,刘 珊,张仟春,*
(1.兴义民族师范学院 生物与化学学院,贵州 兴义 562400;2.兴义民族师范学院 黔西南州食品和环境污染物分析重点实验室,贵州 兴义 562400)
0 引言
甲醛(Formaldehyde,FA)是许多工业过程的化学原料,不仅可作为木质材料的粘合剂,还可作为防腐剂、杀菌剂、消毒剂、生产肥料、纸张和化妆品等[1]。然而,FA却易挥发,接触后会刺激皮肤、喉咙、肺和眼睛等,进而引发头痛、腹痛、呼吸困难、呕吐、鼻咽癌、白血病等各种健康问题,过量摄入还会导致死亡[2]。鉴于此,国际癌症研究机构将其列为1类致癌物清单[2],欧盟委员将其列为1B类致癌物和诱变剂[3]。为更好地对FA的排放进行监管,一些学者对其的吸附行为进行了相关研究[4-6]。
石墨烯(Graphene,G)是一种碳原子以sp2方式杂化方式排列成六边形呈蜂巢晶格的二维碳纳米材料,G由于其出色的机械、良好的导热性、大的比表面积、高的载流子迁移率、良好的导电性和高效吸附性等而引起了研究者的极大兴趣[7-13]。2007年,Geim等[7]首先使用了G用于气体吸附,发现G纳米材料可用于检测单个气体分子。随后,有研究显示,本征G对大多数气体分子的物理吸附较弱,将缺陷和其他金属或非金属原子引入G,可以有效地加强G与气体分子之间的电荷转移,从而提高对某些气体分子的吸附能力[14-17]。在实验和理论计算中,也有学者们试图通过引入某些缺陷或金属原子负载来增强气体分子与G的相互作用。诸如,Wang等[18]基于第一性原理计算发现空位的存在不仅有助于过渡金属原子的吸附,而且还有助于CO气体分子的吸附;Sanyal等[19]采用密度泛函理论(Density functional theory,DFT)计算发现双空位缺陷的存在可以促进O2等气体的吸附;Zhang等[14]采用第一性原理计算发现Au修饰G利于吸附H2S等气体分子;Liang等[16]采用DFT研究发现Ga掺杂可以有效增强其对H2O等气体分子的吸附能力;Shukri等[17]使用第一性原理计算发现掺杂Pd可以增强G吸附CO等气体的能力;Singla等[20]根据DFT计算发现Cu和Pt掺杂可以提高对H2分子的吸附能力。
基于上述研究,本研究通过DFT理论研究了本征石墨烯(Intrinsic graphene,IG)和Au掺杂G(Au-doped graphene,Au-G)对FA分子的吸附特性。首先,建立了FA分子、IG、Au-G的几何模型;其次,在这些模型上优化了不同角度的FA分子的吸附构型,以了解FA分子的最佳吸附形式;最后,通过分析吸附能、差分电荷、电荷转移、态密度及化学键变化的差异,比较了2种模型对FA分子的吸附能力。这些研究可为寻找对FA气体具有高灵敏度的理想传感材料提供理论参考。
1 方法与模型
研究中的理论计算均采用基于DFT的VASP(Vienna Ab-initio simulation package)软件包来进行计算[21-22]。赝势采用投影缀加平面波,交换关联采用的是广义梯度中的PBE (Perdew-burke-ernzerhof)泛函[23]。电荷转移量使用Bader软件进行分析。
计算过程中,采用DFT+U的方法对Au的 4d 轨道进行修正,U值为7.0 eV,平面波的截断能为400 eV,自洽迭代的能量收敛标准为10-5eV,几何优化过程中每个原子受力收敛标准为0.02 eV/Å。IG模型是通过去除石墨晶胞(MS中自带的graphite.msi)对称性并删除一层原子建4×4×1的超胞获得(包括32个碳原子),z方向上真空层厚度取15 Å。采用Monkhorst-Pack方法进行布里渊区数值积分计算,k点网格取值为6×6×1。Au-G模型的构建是将IG模型中的1个C原子替换为1个Au原子。
吸附能的计算公式如下:
Eads=Etotal-Egraphene system-Emolecule
(1)
式(1)中Etotal为FA吸附IG基底(或FA吸附Au-G基底)的能量,Egraphene system为IG基底(或Au-G基底)的能量,Emolecule为单个FA分子的能量。
差分电荷密度的计算如下:
Δρ=ρtotal-ρgraphene system-ρmolecule
(2)
式(2)中ρtotal为吸附态的电荷密度,ρgraphene system为吸附态中IG基底(或Au-G基底)的电荷密度,ρmolecule吸附态中分子的电荷密度。使用vaspkit软件对电荷进行差分和态密度处理[24]。
2 结果与讨论
为分析FA分子吸附在IG和Au-G基底上的吸附能、磁矩变化、电荷转移、态密度和化学键。首先分别对FA分子、IG、Au-G进行结构优化,在优化后的IG和Au-G基底上充分考虑FA分子的可能吸附位点,基于此构建相应的吸附模型,然后进行结构优化,在同种基底吸附FA分子的不同模型中,其能量最低的模型为最稳定构型。
在优化结构图1(a)观察到Au原子与G有1个吸附高度(h),这是因为Au原子半径比碳原子半径大,h计算结果显示为1.719 Å。而Au-C键的键长为2.081 Å,与已有研究一致[25]。图1(b)为Au-G的差分电荷密度图,观察到的电荷再分配主要发生在Au-C键周围,表明形成了强共价键[26]。
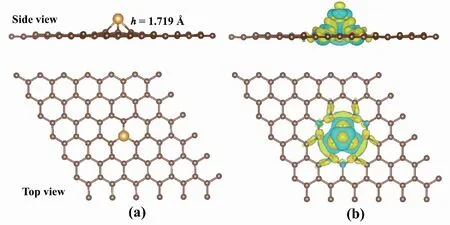
图1 Au-G的结构优化和差分电荷密度Fig.1 The optimized structure and charge density difference of Au-G注:(a)为Au-G结构优化,(b)为Au-G差分电荷密度;黄色代表电荷密度增加,蓝色代表电荷密度减少,电荷等值面为0.001 e/Bohr3。
从图2(a)可看出,自旋向上和自旋向下DOS曲线不对称,表明Au-G具备磁性,通过计算IG和Au-G的磁矩,发现IG的磁矩为0 μB,而当掺杂Au原子后,磁矩变为1.017 μB;图2(b)为Au-C中Au原子的5d轨道及相邻碳原子的2p轨道上的投影态密度(Partial density of states,PDOS),Au-G的磁矩是来源于金原子的5d轨道和相邻碳原子2p轨道上自旋电子的耗损[26]。采用Bader软件分析电荷,结果显示Au原子掺杂在G过程中失去0.183 9 e的电荷,表明Au原子的电荷部分转移到G上。
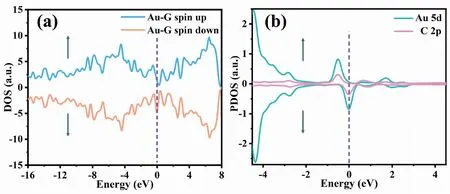
图2 Au-G的总态密度和Au原子的5d轨道及相邻C原子的2p轨道上的投影态密度Fig.2 DOS of Au-G and PDOS in the 5d orbital of Au and the 2p orbital of adjacent C注:(a)为Au-G的总态密度,(b)为Au原子的5d轨道及相邻C原子的2p轨道上的投影态密度。
图3为FA分子吸附在IG和Au-G基底上的最稳定构型。图3(a)显示,FA分子平行吸附在IG基底上的高度为3.431 Å;图3(b)所示,FA分子吸附在Au-G上时,吸附高度为1.607 Å,明显低于吸附在IG上的高度。图4为FA分子吸附在IG和Au-G基底上的差分电荷密度图。图4(a)所示,FA分子与IG基底之间仅有少量电荷从IG基底向FA分子转移,而从图4(b)中观察到FA分子周围的电荷密度明显增加,且FA分子中C原子和O原子之间的电荷密度减少。此外,Au-G的磁矩为1.017 μB,但在吸附FA分子后磁矩明显降低,表明Au-G可通过分子吸附来调节其磁性。以上结果说明FA分子和Au-G之间发生了复杂的电荷转移,表明Au掺杂了G后,其与FA分子的相互作用明显增强。
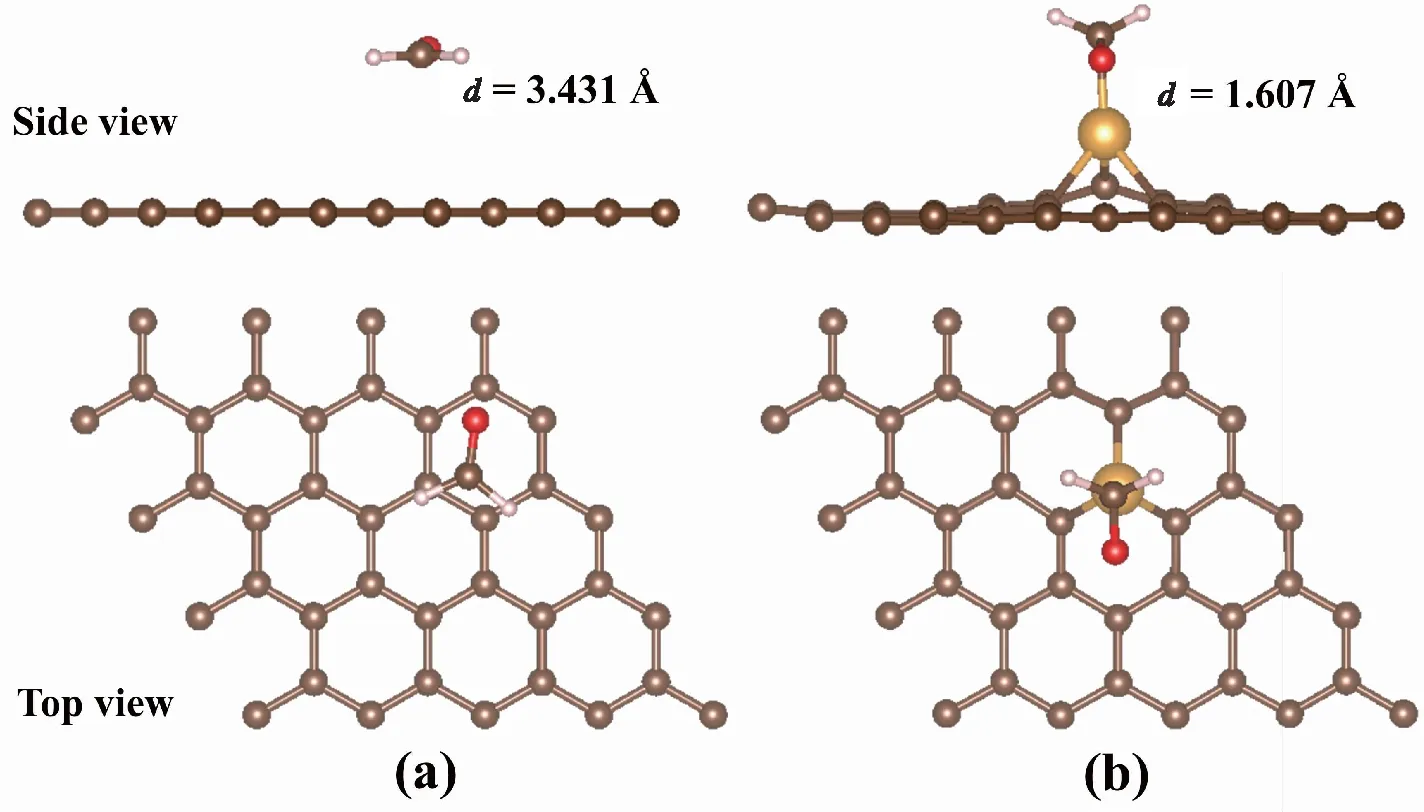
图3 FA吸附在IG和Au-G上的结构优化Fig.3 The optimized structure of FA molecule adsorption for IG and Au-G注:(a)为FA吸附在IG的结构优化,(b)为FA吸附在Au-G上的结构优化。
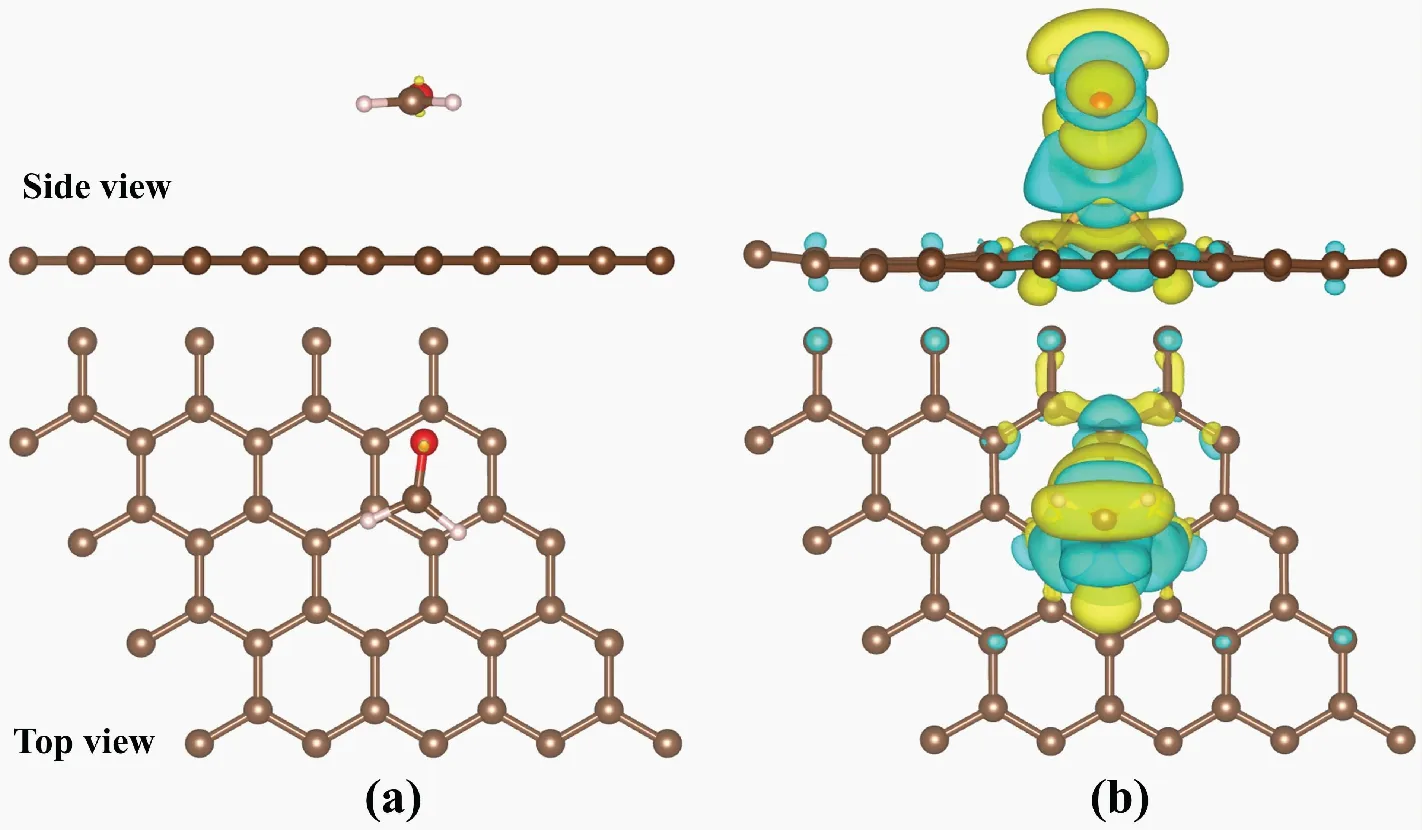
图4 IG和Au-G吸附FA的差分电荷密度Fig.4 The charge density difference of IG and Au-G注:(a)为FA吸附在IG上的差分电荷密度,(b)为FA吸附Au-G上的差分电荷密度;黄色代表电荷密度增加,蓝色代表电荷密度减少,电荷等值面为0.001 e/Bohr3。
表1中的物理量Eads表示FA分子吸附IG和Au-G的吸附能、d表示FA分子吸附在IG和Au-G基底上的吸附高度、dO-Au表示FA分子中O原子与Au-G中Au原子之间的键长、dC-H表示FA分子中C原子与H原子之间的键长、dC-O表示FA分子中C原子与O原子之间的键长、ΔQ表示吸附时从基底向FA分子转移的电荷量、Mag表示吸附前FA分子或不同基底的磁矩、Mag'表示FA分子吸附在不同基底上的磁矩。
由表1数据可知,FA分子吸附在IG和Au-G基底上的吸附能Eads分别为-0.035 eV和-1.218 eV,与IG相比,Au原子的掺杂增大了其对FA分子的吸附能。通过Bader软件分析发现,IG吸附FA分子时IG基底向FA分子转移的电荷量为0.008 4 e,FA分子吸附IG前后C-H键和C-O键键长不变,表明FA分子吸附IG是属于较弱的物理吸附,且FA分子容易从IG表面脱附,说明FA分子不宜吸附到IG表面;此外,通过Bader软件分析发现,Au-G吸附FA分子时Au-G基底向FA分子转移的电荷量为0.376 4 e,Au-G吸附FA分子后FA分子中C-O键键长增长0.123 Å,表明Au原子的掺杂削弱了FA分子中的C-O键。

表1 FA分子吸附在不同G基底结构上的优化参数Tab.1 The optimized parameters of FA molecule absorbed on different G substrates
图5(a)显示,Au-G吸附FA分子前,自旋向上和自旋向下DOS不对称,说明吸附前Au-G基底具备磁性,但吸附FA分子后,自旋向上和自旋向下的DOS几乎对称,表面吸附FA分子后几乎不具备磁性,且Au-G吸附FA分子后使费米能级附近态密度发生明显变化,在偏离费米能级左右两侧-0.666 4 eV和0.467 2 eV新增自旋向上和自旋向下的波峰;由图5(b)可知,-0.666 4 eV位置的波峰主要是由Au的6s轨道和O的2p轨道杂化而成,而0.467 2 eV位置的波峰则是由Au原子的5d轨道、6s轨道及FA分子中C原子的2p轨道和O原子的2p轨道共同杂化而成,这是由于Au原子的6s轨道与FA分子中C原子的2p轨道和O原子的2p轨道发生了杂化从而导致Au原子周围电荷重新分配,并有效的消除了未成对电子,表明Au-G对FA分子具有较好的敏感性。
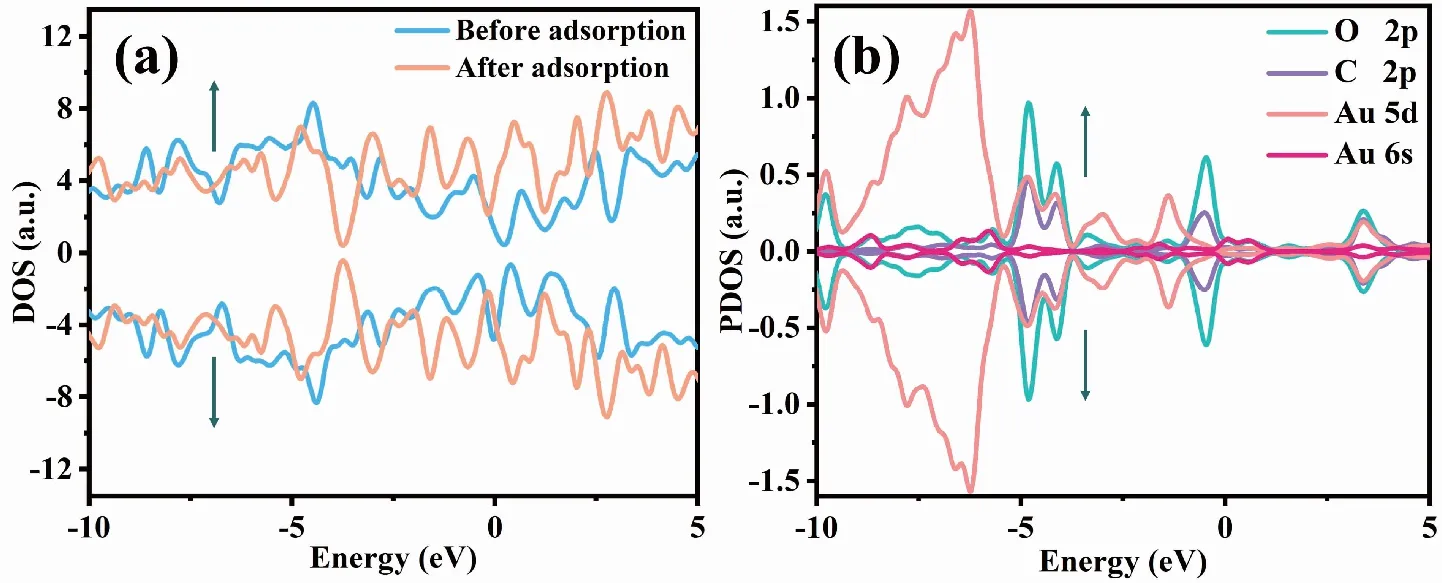
图5 Au-G在吸附FA分子前后的总态密度及吸附体系中Au原子5d轨道和6 s轨道、FA分子中O原子2p轨道和C原子的2p轨道上的投影态密度Fig.5 DOS of Au-G before and after adsorption of FA molecule,and PDOS on the 5d orbital and 6s orbital of Au atom in the adsorption system,2p orbital of O atom and 2p orbital of C atom in FA molecule in the adsorption system注:(a)为Au-G在吸附FA分子前后的总态密度,(b)为吸附体系中Au原子5d轨道、6s轨道及FA分子中O原子2p轨道和C原子的2p轨道上的投影态密度。
通过COHP方法[27-28]进一步探究FA分子与IG和Au-G基底间化学键的变化,用LOBSTER软件进行COHP分析[29-30],在投影过程中基组使用pbeVaspFit 2015[30]。图6中列出的ICOHP值对应相应颜色原子之间的相互作用或轨道对这些相互作用的贡献,图6(a)显示孤立FA分子中C-O键的ICOHP为-7.92 eV;图6(b)观察到FA分子吸附在IG基底上后,FA分子吸附后与IG之间的C1-C2和C2-O的ICOHP分别为0.00 eV、-0.01 eV,FA分子中C-O之间的ICOHP由吸附前的-7.92 eV变为-8.30 eV,这说明FA吸附在IG基底上后未形成化学键,且吸附以后FA分子中C-O键略微被增强;图6(c)和(d)显示,FA分子吸附在Au-G基底上后,C1-Au之间ICOHP为-1.03eV(其中5d-2p和6s-2p对成键的贡献最大),O-Au之间的ICOHP为-0.78 eV(其中5d-2p和6s-2p对成键的贡献最大),FA分子中C-O之间的ICOHP由吸附前的-7.92 eV变为-6.30 eV,这表明FA吸附在Au-G基底上后形成了较弱的C1-Au键和Au-O键,且吸附以后FA分子中C-O键被削弱。
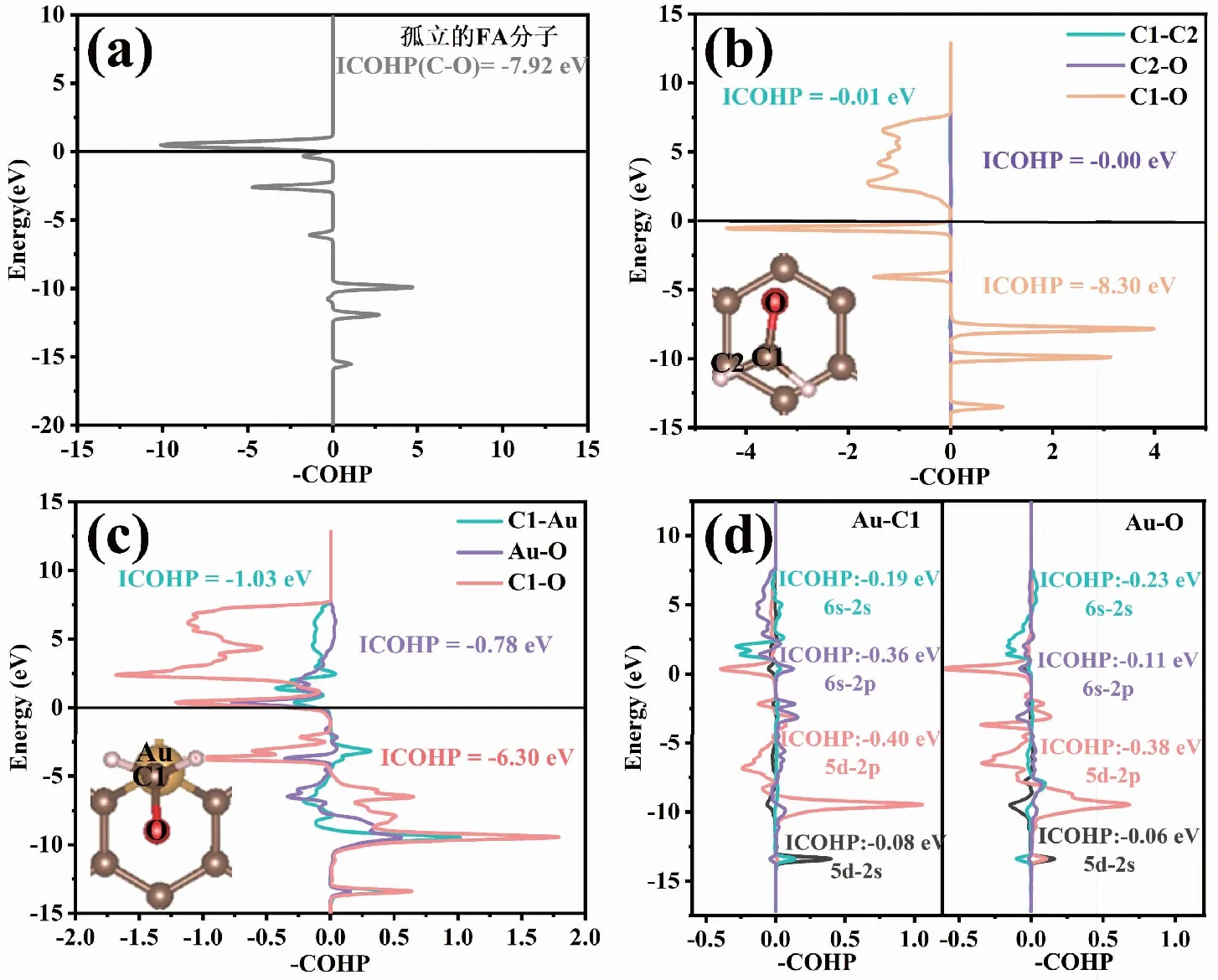
图6 孤立的FA分子及FA分子吸附在IG和Au-G上的COHP分析Fig.6 COHP analysis of isolated FA molecules and FA molecule adsorbed on IG and Au-G 注:(a)为孤立的FA分子的COHP分析,(b)为FA分子吸附在IG上的COHP分析,(c)为FA分子吸附在Au-G上的COHP分析,(d)为FA分子吸附在Au-G上Au-C1和Au-O的COHP分析。
3 结论
采用基于密度泛函理论的第一性原理计算方法,讨论了本征G和Au掺杂G对FA分子的吸附特性。结果表明:FA分子吸附在本征G基底上,吸附能、磁矩变化和电荷转移量分别是-0.035 eV、0.0 μB和0.008 4 e,FA分子吸附在Au掺杂G上,吸附能增加至-1.218 eV,磁矩变化增大到1.012 5 μB,电荷转移量增至0.376 4 e,态密度分析结果说明FA分子与Au掺杂G基底之间存在轨道杂化,且COHP分析结果说明FA分子吸附Au掺杂G上后形成了较弱的Au-O键和Au-C键,表明Au掺杂G对FA分子具有更强的吸附作用。本研究结果可为开发一种高灵敏度探测FA分子的新型传感材料提供理论依据。
