铁离子失衡在阿尔茨海默病发病机制中的作用研究进展
2022-12-26郭爽陈凤燕尹香王璐郭雪峰余启明邹珍友舒伟
郭爽,陈凤燕,尹香,王璐,郭雪峰,余启明,邹珍友,舒伟*
晚期阿尔茨海默病(AD)患者的认知、情绪等心理活动异常,心理健康状况不佳,身体功能也逐渐丧失[1]。随着中国社会老年人口的快速增长,中国AD患者的数量正在迅速增加。然而,AD的发病机制尚不明确,能够完全治愈AD或缓解症状的药物尚未开发。因此,进一步了解诱发或促进AD的病理因素至关重要。
目前研究表明,基于观察到AD患者大脑皮质、皮质下和白质区域的铁含量升高,推测铁离子负载过度可能在AD病理发展中有重要作用。海马区铁水平的增加是AD早期的一个重要特征,与记忆力水平呈负相关[2]。大脑中铁负荷增加会加速β-淀粉样蛋白(Aβ)斑块形成和过度磷酸化的Tau蛋白(pTau)缠结的聚集,并增加其毒性[3]。铁、淀粉样变性和AD之间的关系是多方面的。本文从4个基本的问题出发,系统地了解铁在AD发病机制中的作用,相信以铁为靶点的研究将为AD的临床研究和靶向治疗提供新的方向。
1 铁稳态失调是AD的主要因素还是次要因素?
随着时间的推移,铁在大脑中积累,在30~40岁达到一个明显的峰值。虽然有研究表明脑铁水平相对稳定,但60岁以后个体之间的脑铁分布存在异质性[4]。脑铁水平相对下降可能反映出大脑铁吸收减少或铁释放增加,脑铁相对增加说明大脑铁吸收增加或铁释放减少。脑铁的增加可能受到以下几个因素的影响,包括铁转运和储存蛋白的不平衡、线粒体功能障碍、神经血管机制以及髓鞘破坏和失修。晚发性AD大多出现在65岁以后,与大脑铁峰值一致,早发性AD最早出现在40岁,与大脑铁水平的峰值相吻合[5]。
AD的两个主要组织病理学特征是细胞外Aβ大量沉积形成老年斑和细胞内积聚异常修饰的pTau形成神经原纤维缠结(NFTs)。基于这些明显的病理特征,学者们对AD的发育机制提出了两种假设:淀粉样级联假说和NFTs假说。淀粉样级联假说源于β-淀粉样前体蛋白(APP)的淀粉样降解途径。这一假设表明,APP淀粉样蛋白降解途径产生的Aβ1~42具有显著的神经毒性,引起神经元损伤,最终导致痴呆。NFTs假说起源于AD患者神经元中过度磷酸化的pTau聚集而形成的大量纤维缠结。这一假设表明,过度磷酸化的pTau与正常的pTau竞争与微管蛋白结合,破坏微管组装和分解的动态平衡,并导致轴突运输障碍和细胞内废物积累,以致神经元逐渐退化,引起痴呆。随着研究的深入,越来越多的学者提出了炎症假说和金属离子假说,不断丰富着AD的发展机制。研究人员发现AD患者大脑中存在大量活化的星形胶质细胞和小胶质细胞,同时伴有肿瘤坏死因子α(TNF-α)、白介素(IL)-1β、IL-6等炎性因子的表达水平升高[6],据此,提出了神经炎症假说。这一假设表明,神经炎症在AD中不是由老年斑(SPs)和NFTs激活的被动系统,而是像斑块和缠结一样,在疾病的发展中起着重要作用。有研究表明,铜、铁、锌、镁、铝等金属离子参与了AD的发生和发展[7]。金属离子可以影响神经元代谢,引起氧化应激,促进Aβ的沉积和SPs的形成。同时,还有研究表明,Aβ在大脑中的沉积及其毒性与铜、铁、锌和其他金属离子在皮质和海马中的代谢紊乱直接相关。此外,金属稳态失衡可直接导致神经元功能障碍,并导致神经元细胞死亡[8]。在上述研究的基础上提出的金属离子假说强调金属离子在AD发病中的作用,进一步补充了AD的发病机制。
金属离子假说强调了衰老过程中金属离子氧化还原循环不当的作用,以及大脑中金属平衡失调是如何导致AD中的淀粉样沉积、细胞功能障碍和凋亡。铁并不是唯一在衰老和病变的大脑状态下稳态失调的过渡金属。铜和锌在大脑中也起着重要的生理功能,但其浓度远低于铁。目前的发现表明,金属平衡应包括在淀粉样级联假说的框架内。APP与AD的关联已经被证实。APP经几种酶处理,产生较小的片段,以一种未知的方式参与正常的细胞功能。APP被α-分泌酶(ADAM)或β-分泌酶(BACE-1)切割,产生细胞外可溶性蛋白sAPPα或sAPPβ片段。α途径是非淀粉样变性过程,而β途径是淀粉样变性过程。膜结合的C端片段,即在α和β裂解过程中产生的C83或C99,分别被γ-分泌酶裂解生成细胞外片段p3或Aβ40/42。Aβ42的增加导致不溶性细胞外淀粉样纤维和淀粉样斑块的形成。Fe2+和Fe3+与APP和Aβ的相互作用促进了淀粉样体聚集的程度和速度[9]。Aβ42和Aβ40均可以与铁相互作用,但Aβ42片段形成淀粉样体聚集的速度更快。此外,APP/PS1AD小鼠模型的体内铁超载导致APP处理增加,神经信号改变,随着淀粉样斑块形成的增加导致认知能力下降[10]。这表明脑铁负荷直接与淀粉样蛋白加工和斑块形成的初始阶段有关。虽然有发现表明AD脑的总铁含量没有改变,但区域分析提供了证据,AD大脑的活体MRI成像显示海马区中的铁含量增加,海马区是大脑中受淀粉样变性影响的中央区域,这也表明铁的沉积发生在淀粉样斑块周围[11]。
因此,铁稳态失调有可能在一定条件下通过促进Aβ形成而导致AD的发展,在某些类型的AD中有可能是AD发生的主要因素。
2 脑铁失衡是源于外周铁还是内源性脑铁?
大脑中的铁不均匀地分布于基底核区的神经元、脑毛细血管内皮细胞(BCECs)和胶质细胞。大脑需要比任何其他器官更多的储备非血液铁来执行其功能。首先,大脑是身体中代谢最活跃的器官之一,消耗人体大量的氧气,这需要大脑有一定的铁储备,以确保在铁状态的潜在静止期满足人体能量需求。其次,铁流入和流出大脑受到血脑屏障(BBB)、脑-脑脊液屏障(BCSFI)和血-脑脊液屏障(BCSFB)的严格控制[12],这种调节存在于大脑的控制之外,使得大脑很难精细地调整铁的流入和流出。再次,虽然成年大脑中神经元的生长和分裂很少,限制新的外源性铁创造新的突触连接,但富含铁的少突胶质细胞持续需要大量的铁元素。
铁在大脑中的稳态平衡是通过6个不同的过程来协调的:运输、摄取、储存、利用、氧化还原循环和排出。铁在全身的大部分运输通过糖蛋白转铁蛋白(TF)进行。少突胶质细胞和脉络丛细胞是产生TF的两种神经细胞。TF在白质中的表达下降,皮质的晕区和周围产生Aβ斑块,表明该组织有损伤。除了分布在少突胶质细胞内,TF还在AD脑的星形胶质细胞中被发现异常,表明这些细胞中存在铁稳态失衡[13]。
脑铁摄取一般由转铁蛋白受体(TFR)1和二价金属转运体1(DMT1)调节。细胞外部分Fe2+可通过DMT1直接转运到细胞内,Fe3+与转铁蛋白结合通过内吞作用与TFR1结合,在细胞内形成内质体。由于质子泵对内质体的作用,Fe3+从TF/TFR1复合物中解离并还原为Fe2+,Fe2+通过DMT1进入细胞质。在BBB的动脉内皮细胞和BCSFB的脉络丛上皮细胞上TFR1高表达,在神经细胞和反应性小胶质细胞表面也有表达,以促进炎症过程中神经元铁的导入和小胶质细胞铁的固存[14]。海马区是一个受早期淀粉样变严重影响的区域,海马齿状回(DG)是AD大脑中神经元和新分裂细胞TFR1高度表达的区域。AD患者在该区域形成Aβ斑块的原因还没有得到彻底的证实,但被假设与该区域无法限制铁的摄入量有关。DMT1促进非转铁蛋白结合铁(NTBI)在运输过程中的摄取,其位于12号染色体上的基因序列(SLC11A2)在AD中常见突变[15]。此外,炎性细胞因子也会增加AD脑神经元、星形胶质细胞和小胶质细胞内DMT1的表达。
细胞铁的储存、解毒和释放是由铁蛋白调节的,H-铁蛋白作为铁氧酶,在铁蛋白的核心将Fe2+氧化为Fe3+并储存。在正常衰老过程中,铁蛋白的分布在整个大脑中保持不变。铁蛋白主要存在于少突胶质细胞中,同时也存在于神经元、原质星形胶质细胞和小胶质细胞中。体内MRI测量表明,与对照组相比,年轻晚发性AD患者的基底核铁蛋白含量更高,而老年晚发性AD患者的基底核铁蛋白含量没有明显变化[16]。AD脑在淀粉样斑块和血管周围呈现出小胶质铁蛋白高表达,脑脊液铁蛋白水平升高在AD中也很明显。
部分进入细胞的Fe2+被线粒体利用,如frataxin、mitoNEET和大脑中需要铁作为电子载体的其他酶。frataxin被认为在神经元对Fe-S簇的组装中起着铁伴侣的作用。此外,最近发现了一种线粒体外frataxin,可以稳定乌头酶中的Fe-S簇,为胞质乌头酶/铁调节蛋白(IRP)1提供分子控制[17]。mitoNEET是线粒体外叶中控制铁的蛋白质。其通过控制导入线粒体的铁量和控制线粒体呼吸产生的氧化物来调节最大呼吸速率。铁基质掺入和氧化应激的增加有效增强了线粒体呼吸能力,进而减少了mitoNEET。据推测,frataxin和mitoNEET的损伤或功能障碍涉及AD的几个方面,包括活性氧(ROS)控制、氧化还原控制和血红素代谢。然而,没有研究表明frataxin和mitoNEET功能障碍与淀粉样蛋白的产生直接相关。
机体需要铁的氧化还原循环满足产生ROS的细胞能量需求。细胞外和细胞内的H-铁蛋白将可溶性Fe2+氧化成不溶性Fe3+来解毒和储存铁。亚铁被铜蓝蛋白和亚铁氧化酶氧化,随后由膜铁转运蛋白和铁转运蛋白以Fe3+的形式排出。体内STEAP3和外周膜细胞朊蛋白(PrPc)还原Fe3+,通过TFR和NTBI促进铁的内流[18]。Aβ与PrPc的结合会损害铁还原酶活性,PrPc的丢失会导致大脑铁超载,同时TF和TFR1转录升高。
铁转运蛋白参与铁的排出。铁转运蛋白与氧化铁酶(如铜蓝蛋白)一起将细胞内的Fe2+转运并氧化为Fe3+。由此产生的Fe3+通过细胞膜的铁转运蛋白1(FPN1)被运出细胞。铁调素主要由肝脏分泌,以控制周围铁水平。如果铁在铁转运蛋白核心中没有被氧化,铁调素就会与铁转运蛋白结合并触发受体配体复合物的内化和降解。在AD小鼠模型中,铁蛋白的减少与受损血管附近的血红素颗粒沉积有关,这表明血管功能障碍改变了AD脑铁的流动和代谢稳定[19]。
铁在大脑中通过运输、摄取、储存、利用、氧化还原循环和排出6个过程维持稳态平衡。脑铁摄取由TFR1和DMT1调节,TFR1在神经细胞和反应性小胶质细胞表面表达,以促进炎症过程中神经元铁的导入和小胶质细胞铁的固存,海马齿状回(DG)是AD大脑中神经元和新分裂细胞TFR1高度表达的区域;DMT1促进NTBI在运输过程中的摄取,炎性因子会促进其表达。少突胶质细胞和脉络丛细胞产生的TF负责全身大部分的铁运输,TF在AD大脑中的表达下降,这表明铁稳态失衡与AD有关。主要存在于少突胶质细胞中的铁蛋白调节细胞的铁储存,脑脊液铁蛋白水平升高在AD中也很明显。Fe2+在线粒体被利用,如frataxin(共济蛋白)可以稳定神经元中的Fe-S簇,mitoNEET可以控制导入线粒体的铁量,线粒体的损伤或功能障碍涉及AD的多个方面。机体需要铁的氧化还原循环满足产生ROS的细胞能量需求。铁过量的氧化还原产生过量的ROS,进而诱发AD(图1)。
另外特品屋还出售西栅老街上各个作坊出产的特产,比如宏源泰染坊将蓝印花布与现代成衣技术相结合,设计出许多很多新潮的衣服,符合现代人的审美同时极富江南特色,也不会让人觉得老土。
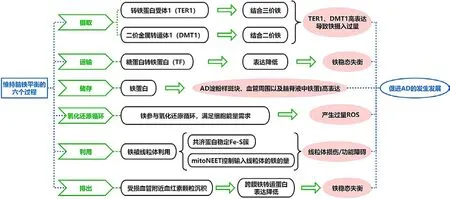
图1 大脑中铁稳态失衡与AD发生发展的关系示意图Figure 1 Schematic diagram of the relationship between the imbalance of iron homeostasis in brain and the occurrence and development of AD
由此看来,脑体失衡在一定程度上发生在外周铁运输、摄取、储存、利用、氧化还原循环和排出这一过程中,任何一个环节的异常均会导致脑铁失衡的发生。
3 AD脑铁失衡是由功能失调还是铁调节系统不堪重负引起?
根据尸检报告,人脑中的总铁沉积与年龄呈正相关,在壳核、苍白球和黑质的基底核区含有高浓度的铁,而大脑皮质、脑干和小脑含有低浓度的铁[20]。神经细胞中的铁稳态主要受参与铁代谢的mRNAs转录水平的调控。参与脑铁代谢的蛋白质主要包括IRP、TF、TFR1、铁蛋白、FPN1、DMT1等。编码TFR1、铁蛋白、FPN1和DMT1的mRNA均含有一种特殊的氨基酸序列,称为铁调节元件(IRE)。铁通过作用于IRE位点来调节铁相关蛋白的转录,从而维持细胞内铁的稳态[21]。低氧诱导因子(HIF)-1和HIF-2的转录调控着所有细胞类型的铁平衡。HIF1和HIF2在结构上相似,但其有独特的糖酵解控制,在大脑离散细胞中表达。HIF由一个胞质亚单位HIFα和一个核亚单位HIFβ组成。HIF异二聚体在低铁、低氧或高一氧化氮(NO)条件下形成,产生含铁调节元件(IREs)的靶RNA转录物,包括TFR1和DMT1。当铁和氧充足时,HIFα的降解由泛素-蛋白酶体系统介导。缺血性事件、铁负荷或蛋白酶体功能紊乱改变了AD患者的HIF功能,降低了TFR1和DMT1水平。有研究用脯氨酰-羟化酶抑制剂稳定正常衰老小鼠的HIF可增强海马记忆,为AD提供了一种潜在的治疗方法[22]。铁螯合剂,如M30,可上调皮质神经元中的HIF和HIF活化化合物[23]。
经过不断深入的研究,人们发现了一种抗菌肽——Hepcidin,其是铁稳态的重要因素,特别是脑铁稳态[24]。FPN1是体内Hepcidin的主要受体。研究表明,Hepcidin通过Hepcidin和FPN1之间的直接相互作用调节铁的稳态,诱导FPN1的内化和降解,降低细胞输出铁的能力,从而增加细胞内铁超载的可能性[25]。细胞实验表明,Hepcidin不仅下调了FPN1的表达,而且还下调了TFR和DMT1的表达[26]。上述结果表明,Hepcidin既通过控制细胞的铁输出,又通过调节细胞的铁输入来调节铁的稳态。
除了调节铁相关蛋白外,APP和pTau还起调节铁的作用。研究表明APP是铁稳态的某种调节因子,其可以与FPN1相互作用,调节铁离子的流出[27]。pTau作为细胞内微管相关蛋白,可将产生的APP转运到细胞表面,促进铁的输出。同时Tau基因敲除小鼠会产生年龄依赖性的铁积累和脑萎缩,铁在原代培养神经元中的滞留是由于APP表面转运减少所致,提示Tau介导的铁稳态可能依赖于APP[28]。
脑铁平衡受到多种调节铁的相关蛋白的调控和APP及pTau对铁的调节作用,因此可以推测,AD脑铁失衡是由功能失调引起的。
4 AD的病因是Aβ斑块形成、铁失稳态,还是两个过程的协同作用?
4.1 Fe与Aβ沉积和τ过渡磷酸化 研究表明,铁代谢紊乱可以诱导Aβ的产生和积累[29],因为铁可以作用于APP mRNA的IRE位点,从而增强内源性APP的翻译和表达。研究人员利用透射电子显微镜(TEM)证实,铁以氧化铁(Fe3O4)磁铁矿纳米粒子的形式存在于SPs的核心中。这为铁积累和Aβ聚集相关提供了金属生物学证据。后来,利用X射线磁性圆二色吸收谱再次揭示了人类SPs中存在磁铁矿。磁铁矿,作为一种多晶氧化铁,不是人脑的正常特征,其含量升高表明异常的铁氧化还原与AD有关。此外,TELLING等[30]利用高级亚显微分辨率X射线显微技术,找到了SPs结构与铁形成淀粉样铁复合体的直接关系。Aβ通过肽的亲水N端区域的3个组氨酸残基和1个酪氨酸残基与铁结合,这有助于稳定这些铁离子。研究还发现,亚铁离子与Aβ的结合降低了肽螺旋结构并增加了肽的β-片段含量,这表明亚铁离子通过增强肽与肽之间的相互作用促进淀粉样单体形成低聚物和原纤维[31]。除促进Aβ聚集外,高铁水平可影响APP的淀粉样变性过程。铁对APP的α-分泌酶切割活性有调节作用。α-分泌酶和β-分泌酶从非活性状态转化为活性状态的过程受呋喃调节,铁可以在转录水平上调节呋喃的表达。过量的铁会抑制有利于β分泌酶激活的呋喃的表达,从而促进淀粉样蛋白途径中Aβ的产生[32]。
体外研究也发现铁可以促进Aβ肽的聚集和细胞毒性的增强。然而,关于铁和Aβ的作用却有不同的看法。有研究发现,Fe2+和Fe3+与APP和Aβ相互作用,促进Aβ聚集成纤维状。Fe2+还可以与Aβ蛋白的氨基酸相互作用,这可能以不同于铜和锌的方式改变淀粉样蛋白的形式。与Aβ结合的Fe3+很容易还原为Fe2+,并增加ROS的产生,从而导致β-分泌酶将Aβ42切割成毒性更强的Aβ低聚物,加速神经元死亡[33]。重要的是,Aβ会损伤线粒体功能,将Fe3+转化为具有氧化还原活性的Fe2+,并诱导氧化应激,从而加重铁超载和AD状况。另一项研究表明,Aβ能显著降低铁的氧化还原能力,这可能表明Aβ在AD发病过程中具有神经保护和金属螯合作用,但在一定条件下会产生毒性[34]。
除了Aβ外,铁还可以与pTau结合,诱导pTau磷酸化,并聚集磷酸化的pTau。NFTs是AD的另一主要病理特征,磷酸化的pTau就是NFTs的主要组成部分。研究发现神经元NFTs中有铁沉积,在AD发病过程中pTau间接参与了脑神经元铁离子的传递[35]。
4.3 Fe与自噬 自噬是一种由溶酶体驱动的高度保守的蛋白质水解系统,可以去除功能失调的细胞器和错误折叠的蛋白质,从而在应对各种不利的环境压力和维持细胞活力方面发挥重要作用。自噬不足可引起细胞内蛋白质积累,从而损害细胞功能,而自噬过度也可破坏细胞微环境,造成细胞损伤。在AD脑中发现了自噬不足和自噬过度,自噬不足可能在AD错误折叠蛋白的积累中发挥更重要的作用,如pTau和Aβ的积累[36]。最近的一项研究表明,细胞内pTau的积累会反过来破坏IST1基因调控的内吞体分选转运复合体(ESCRT)-Ⅲ复合物形成和自噬体-自溶体融合,加重自噬缺陷,从而在AD中形成pTau积累和自噬缺陷之间的恶性循环[37]。泛素蛋白酶体系统(UPS)在AD脑中的活性降低,UPS活性与自噬功能密切相关。铁离子可以与UPS组分结合,改变自噬过程。因此,自噬应该是AD的一个有前景的治疗靶点。铁的摄取和铁相关脂褐素的溶酶体积累诱导自噬缺陷。富含铁的自体溶酶体可以产生更多的ROS,从而对自体溶酶体膜造成损害[38]。铁蛋白是一种储存铁的细胞内蛋白质,因此与自噬缺陷相关的铁蛋白升高在AD中积累铁和介导铁毒性方面起着重要作用。同时,细胞质中的铁含量增加会使铁蛋白饱和,阻止铁蛋白进入自溶体,从而加剧铁的积累。
4.4 Fe与突触 突触损伤是AD的早期病理特征,与AD的认知功能障碍呈正相关。铁离子的稳态对维持突触功能至关重要。据报道,膳食铁是突触形成所必需的,缺铁会减少果蝇生物钟回路中突触的形成[39]。铁介导抗N-甲基-D-天冬氨酸受体(NMDAR)依赖性刺激钙诱导通路和海马突触的可塑性。NTBI增加“氧化张力”,这对基础突触传递和长期增强均很重要。在铁含量升高的情况下,突触活性的增加会导致铁超载,这与细胞毒性效应相一致。另一方面,长期过量铁暴露会导致小鼠学习和记忆障碍,包括突触和记忆相关蛋白的广泛分子改变[40]。
4.5 Fe与脑淀粉样血管病变 AD小鼠模型脑微血管铸型显示微血管结构发生改变,并伴有Aβ的附着和沉积[41]。CAA导致Aβ在脑膜和脑内血管沉积。CAA在超过50%的老年人中出现,在50%的AD病例中也有发现,这突出表明CAA在衰老的大脑中很常见,但并不总与AD相关。伴和不伴有CAA的AD患者组织铁负荷存在差异;伴有严重CAA的AD患者的组织中非血红素铁显著升高,尤其是在大血管壁[42]。由此推测Aβ清除不当和BBB铁的积聚可导致异常血管生成和脑血管系统的衰老。这些环境条件开启了神经血管解耦、血管退行性变、血管平滑肌丢失、脑灌注不足和神经血管炎症的过程。最终,这些情况导致BBB渗漏、引流不当、神经元环境中的化学失衡以及突触功能障碍和丢失。淀粉样蛋白沉积的来源是异质性的,目前尚不清楚红细胞Aβ水平如何与CAA相关。然而,红细胞功能障碍有助于解释AD闭塞血管的低灌注和血红素沉积。
综上所述,铁稳态失衡参与多重机制影响AD的发生和发展,同时也促进Aβ的形成与集聚,因此AD的病因是Aβ斑块形成与铁失稳态失衡这两个过程的协同作用。
5 小结
AD的发病机制是异质性的,有许多与其进展相关的生理特征。老化的AD大脑可能吸收更多铁,运输铁不当,或释放铁不足,导致铁离子负荷。铁水平的增加通过调节特定的蛋白激酶或β-、γ-、α-分泌酶途径提升APP的表达,进而通过多种途径促进Aβ和pTau过度磷酸化的形成,诱导或加剧Aβ和过度磷酸化的pTau聚集。与此同时,铁过量引起氧化应激,通过Fenton等反应产生ROS,过量的ROS会导致神经元膜脂质过氧化、DNA损伤和脑内神经元损伤等级联病理反应。铁负荷也通过作用于自噬相关蛋白来促进或抑制自噬,从而引起自噬损伤,诱导自噬缺陷。铁离子的突触释放对正常的突触可塑性和功能至关重要,而其异常的分布或代谢在AD的发展过程中会引起突触功能障碍,其机制与铁离子在大脑不同区域的沉积损害了线粒体的功能密切相关。
长期过量铁暴露会导致突触损伤,这与AD的认知功能障碍呈正相关。过量的铁通过激活CDK5/p25复合物和GSK3β促使pTau的磷酸化进而形成NFTs。铁过量还可以作用于APP mRNA的IRE位点,增加内源性APP的表达,促进Aβ的形成。大脑中的铁代谢异常时,氧化应激通过Fenton反应产生的过量ROS会导致神经元膜脂质过氧化、DNA损伤和脑内神经元损伤进而引发AD。脑淀粉样血管病(CAA)和铁沉积可能导致Aβ在脑膜和脑内血管沉积。过量的铁也会抑制有利于β分泌酶激活的呋喃的表达,从而促进淀粉样蛋白途径中Aβ的产生。泛素蛋白酶体系统(UPS)活性与自噬功能密切相关,在AD脑中UPS活性降低,自噬不足可引起细胞内蛋白质积累,从而损害细胞功能,而自噬过度也可破坏细胞微环境,造成细胞损伤(图2)。

图2 铁离子通过多种途径参与AD的发病机制示意图Figure 2 Schematic diagram of iron ion involved in the pathogenesis of AD through various ways
由于铁超载在AD的发生发展中起着重要作用,因此利用金属螯合剂来减少AD患者大脑某些区域的过量铁,缓解甚至治疗AD的策略受到越来越多的关注。金属螯合剂要有效的发挥螯合作用,必须具有以下特点:(1)易于穿透细胞膜和BBB;(2)靶向铁富集区,而不消耗血浆中转铁蛋白结合铁;(3)将螯合铁从铁累积部位去除或转移到其他生物蛋白,如循环转铁蛋白;(4)对身体无不良反应或轻微不良反应[43]。因此,近年来,许多人试图寻找金属螯合剂作为候选药物,但很少有螯合剂被合理设计用于制造安全有效的药物。氯喹诺尔(CQ)是一种有效的金属螯合剂,CQ与铁、锌和铜离子的高结合亲和力使其能够竞争性地从Aβ中捕获这些金属,并防止Aβ的聚集。去铁胺(DFO)是一种经充分证实的铁螯合剂,可抑制铁或铝的毒性及其在体内诱导的ROS。DFO可以抑制Aβ1~42的形成,并溶解预先形成的淀粉样斑块,同时可以抑制pTau的磷酸化。但DFO的临床应用仍存在许多问题,如:生物利用度差、不能口服、必须注射及存在不良反应。以铁为靶点研究AD的发病机制已成为近年来的研究热点。利用铁螯合剂螯合AD患者大脑中多余的铁也成为治疗AD的新策略,然而,与其他组织器官不同的是,大脑BBB对药物进入具有严格的特异性。因此,必须从人体自身合成的物质中寻找一种更容易穿过BBB并能有效发挥作用的铁螯合剂,α-硫辛酸(LA)和乳铁蛋白(LF)就是这些候选药物之一。LA是一种小分子化合物,是存在于线粒体中的辅酶,容易穿过BBB。LA可能通过激活乙酰胆碱转移酶(ChAT)增加乙酰胆碱的表达,改善AD患者的认知功能。LA还可以发挥抗炎、抗氧化和活化葡萄糖摄取和利用的作用。LA的慢性治疗有效地抑制了pTau过度磷酸化,减轻了神经元的变性和异常行为。LF是一种非血红素铁结合糖蛋白,可以与铁、锌、铜或其他金属离子可逆地结合,并通过血液循环输送到全身组织参与代谢,最终在尿液中排泄。近年来LF被广泛用作药物靶向大脑的载体。LF可能通过抗炎,调节免疫,螯合金属离子,调节细胞自噬等作用来调节Aβ和NFTs的产生,最终可能影响AD的发生和发展。体外和体内实验均证实,直接给予LA和LF可缓解AD症状,但其相关作用机制尚未阐明,有待进一步研究。同时,基于LF的纳米药物分子也在研究中。这种药物分子附着在LF分子上,很容易穿过BBB,从而对病变起到治疗作用,达到更好的治疗效果。
作者贡献:郭爽、舒伟进行文章的构思与设计,文章的可行性分析,文献/资料收集、整理,撰写论文;郭爽、陈凤燕、尹香、王璐进行论文及英文的修订;郭雪峰、余启明、邹珍友、舒伟负责文章的质量控制及审校,对文章整体负责,监督管理。
本文无利益冲突。
