一种基于深度学习的蛋白质组分析方法
2022-12-24刘扣龙郑浩然
刘扣龙 郑浩然
0 引言
近年来随着质谱技术的发展,以及仪器精度的提高,主要采用液相色谱-串联质谱技术(liquid chromatography-tandem mass spectrometry,LC-MS/MS)对大规模蛋白质组进行分析[1]。蛋白质组数据采集策略主要有两种,一种是数据依赖性采集(data dependent acquisition,DDA)[2],另一种是数据非依赖性采集(data independent acquisition,DIA)[3]。在DIA中,将质谱整个扫描范围分为若干个窗口,循环地对每个窗口中的所有离子进行碎裂,而不是选择具有特定质荷比的离子,具有高通量、可重复性高的优点[4]。但采用DIA方法进行碎裂,会导致二级质谱是由多个母离子同时碎裂产生的混合质谱,母离子与碎裂子离子之间不存在对应关系,显著增加了肽段定性和定量的复杂度。
目前在DIA数据的蛋白质组分析中,主要都是基于提取离子色谱图(extracted ion chromatogram,XIC)的方法[5]。例如,OpenSWATH[6]通过集成各种软件工具来辅助DIA分析,提取目标肽的色谱图,对碎片离子的共洗脱峰进行评分,最后进行统计分析,其在数据处理方面较繁琐。Wang等[7-8]计算了实验质谱与理论质谱之间的余弦相似度,针对两个肽段构成的混合质谱的情况进行求解,并给出非混合质谱与混合质谱的区分方法,提高了搜索质谱库的灵敏度。MSPLIT-DIA[9]计算归一化点积,作为图谱之间的相似度,结合色谱峰形、保留时间等相关特征来鉴定肽段。Specter[10]是在上述工作上进行了扩展,将DIA中混合二级质谱的强度看作是不同肽段碎片离子强度的线性叠加,将混合二级质谱和匹配到的肽段质谱进行线性拟合,再将求解的肽段系数构建色谱峰,提取峰特征,可以准确地鉴定出相应的肽段并进行定量分析,但线性求解过程中并不能完全拟合,存在很多误差。使用神经网络提高定性效果的研究,例如DIA-NN[11]在定性时先构建色谱峰,提取色谱峰相关的特征,用神经网络迭代寻找最佳的洗脱峰,从而获取定性结果;定量时使用洗脱峰的积分结果,再进行校正处理,增加了定性和定量的肽段数量。但该方法在定性和定量时依然基于离子色谱图的方式,流程复杂,结果会受到色谱图复杂度和色谱时间的影响。FIGS[12]利用不同肽段质谱中特有的峰对混合二级质谱进行线性拟合,迭代求解每个肽段的系数,再构建色谱峰,进行定性和定量,显著提高了肽段定性和定量的准确度,但该方法同样存在求解时不能完全拟合,以及构建色谱峰时存在误差等问题。
这些基于离子色谱图的方法都需要构建离子色谱峰,经过特征提取、积分等操作,会受到色谱维度的影响。色谱复杂度不同会对离子匹配和构建出的色谱峰形产生影响;而色谱时间的长度和偏移会对离子间的色谱峰相关性产生影响,这些复杂流程中存在很多误差,导致定性和定量结果不准确。针对该方法存在的问题,课题组没有使用色谱维度的信息,不需要构建离子色谱峰,结合深度学习在分类和预测问题上的优势,提出了基于卷积神经网络(convolutional neural network,CNN)的定性和定量模型,通过二分类和回归预测的方式,直接获取肽段的定性和定量结果,从而在减少色谱维度信息影响的同时,有效地进行蛋白质组分析。
1 方法
1.1 预处理
混合二级质谱和肽段的质谱数据特征如图1所示,横轴为m/z,纵轴为峰强度。将肽段对应的质谱峰强度,转换成一维向量形式,同时将肽段和多个混合二级质谱进行峰匹配,将匹配后的峰强度转换成多个一维向量形式,然后,经过预处理和特征提取后输入到CNN中。
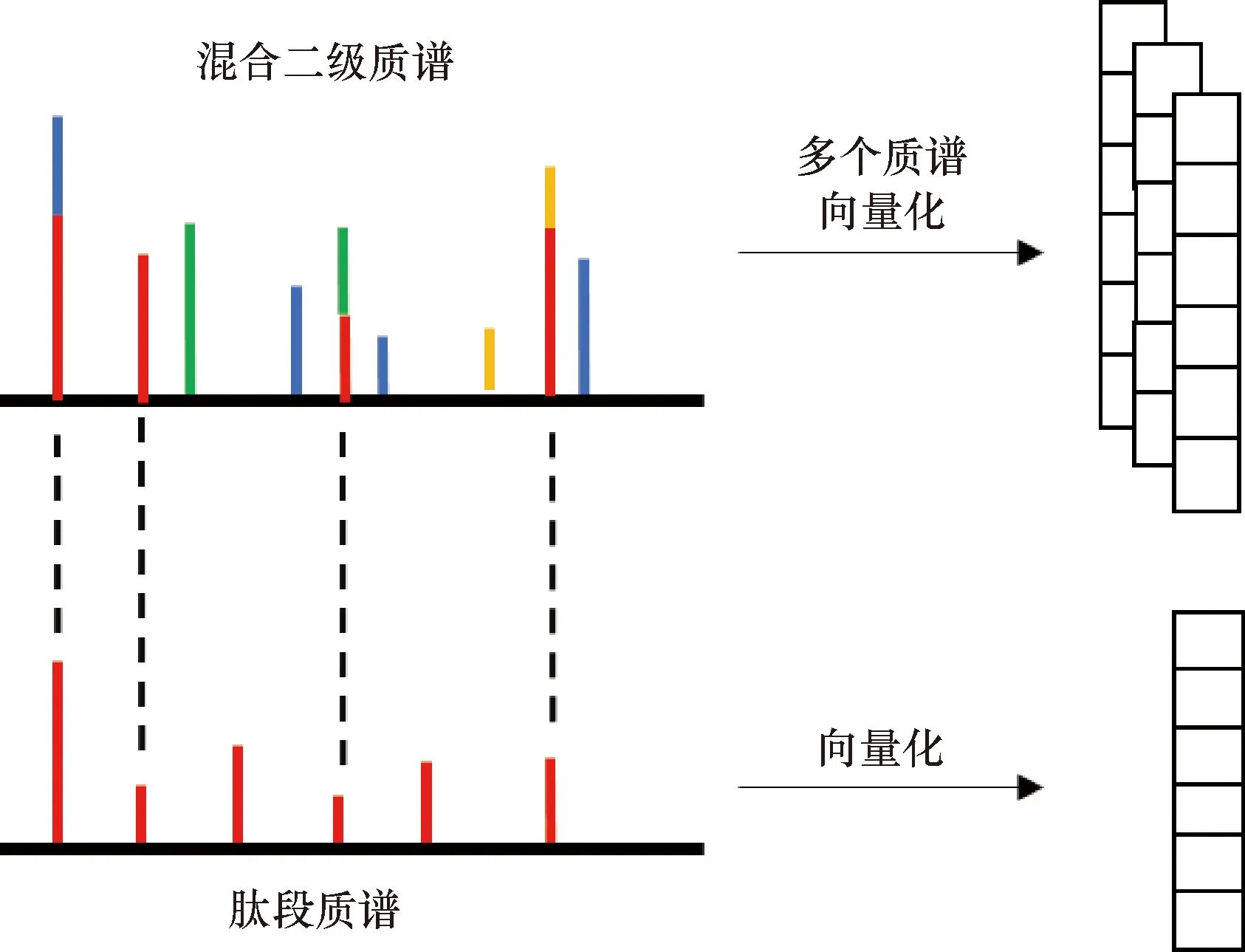
图1 质谱数据示意图Figure 1 Schematic diagram of mass spectrometry data
对于质谱库中的每个肽段母离子,记为Libi,在不同扫描时间点,都可能参与构成其所在扫描窗口对应的每个混合二级质谱,为了找到和Libi最相关的混合二级质谱,采用两个条件进行过滤。
(1) 对于Libi的每个峰对应的m/z值,匹配扫描窗口内的每个混合二级质谱,找到有峰重合的,并且重合数量大于5个的混合二级质谱,仅保留混合二级质谱中与Libi重合的峰。
(2) 将过滤后的每个混合二级质谱,计算其和Libi的相关度,保留相关度最高的s个混合二级质谱(不够s个则用0填充)。使用2个信息进行相关度计算。
第1个信息:计算Libi和匹配到的混合二级质谱的相似度。每个肽段母离子对应质谱的峰强度经过归一化(强度和为1),先对匹配到的混合二级质谱的峰强度做同样的归一化,记为MS2k。这样二者的峰强度就都处于0到1之间了。理论上,对于其中一个混合二级质谱MS2k,如果完全由肽段母离子Libi碎裂形成,即没有其他肽段母离子的成分,则MS2k和Libi对应m/z位置的归一化后的峰强度应该相同。所以计算Libi和MS2k的质谱峰强度差的绝对值,然后求和作为二者间的距离,即:
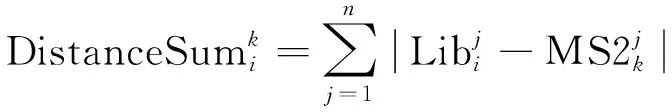
(1)
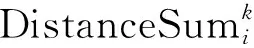
第2个信息:肽段母离子Libi和匹配到的混合二级质谱MS2k,理论上二者越相关,即如果该混合二级质谱MS2k完全由肽段母离子Libi碎裂形成,则MS2k中和Libi对应的峰的强度之和应该越大。所以计算MS2k中和Libi对应的质谱峰的强度之和,作为二者间的距离,即:
(2)
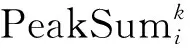
为了同时使用这2个信息,对第2个信息进行处理,把Libi匹配到的混合二级质谱计算得到的PeakSum,除以其中的最大值,这样范围处于0到1之间,与第1个信息的量级一样,然后取负值和第1个信息相加。这样得到的值越小,说明肽段母离子Libi和混合二级质谱MS2k越相关。最后选择最相关的s个混合二级质谱保留下来。
1.2 定性模型
肽段定性需要使用前面预处理后的数据,利用肽段母离子的质谱和该肽段匹配到的混合二级质谱来判定该肽段母离子是否在实验样品中。设计1个基于CNN的二分类模型,若肽段属于该样品,则模型输出的分数接近于1,否则接近于0。这里采用CNN模型,是考虑到输入的质谱数据类似于彩色图片的多通道,并且相邻质谱峰之间存在相关性。而传统的机器学习模型需要提取大量相关的特征,并且会损失原始数据的信息,所以使用CNN,可以更好地提取深度特征。
利用前面预处理后的数据,进行特征提取。主要提取了2个特征,与预处理的相似度类似,但提取的是m/z维度的特征,没有构建色谱峰特征。
(1) 计算肽段匹配到的混合二级质谱的m/z维度的峰强度的和,即将这些匹配到的混合二级质谱合并为1个。
(3)
式中:s表示Libi匹配到的混合二级质谱的个数;j表示Libi和MS2k对应的第j个峰。
(2) 计算Libi和MS2k的质谱峰强度差的绝对值。
(4)

模型结构如图2所示,首先将提取的特征输入到CNN中去,再将提取的深度特征拼接到一起,然后经过全连接层处理,最后经过Sigmoid函数,获取分类属于正样本的概率。使用二元交叉熵作为损失函数:
Loss=-[ylog2p+(1-y)log2(1-p)]
(5)
式中:y为真实标签,值为0或1;p为预测值,范围是(0,1)。
网络结构参数如表1所示。卷积层的输出维度为:[batch size,卷积核个数,峰个数],卷积层的参数个数等于卷积核的个数乘以核的大小。使用Adam算法优化网络参数。

表1 定性模型网络参数Table 1 Network parameters of qualitative model
1.3 定量模型
定量模型的流程和定性模型类似,但是每一步的处理方式不同。肽段定量同样利用前面预处理后的数据,这里不对该数据做其他处理,使用原始数据不会损失任何重要的信息,这样能保证定量预测结果的准确性。设计一个基于CNN的回归模型,直接预测输出该肽段的定量值。
模型结构如图3所示,将肽段质谱和预处理后匹配到的混合二级质谱分别输入到CNN中去,将提取的特征拼接到一起,再经过第二层CNN和全连接层处理,最后输出一个值,作为肽段的定量值。使用均方误差作为损失函数,即:
(6)

图2 深度学习定性模型结构图Figure 2 Structure diagram of deep learning qualitative model
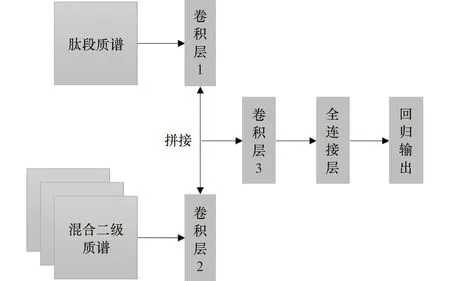
图3 深度学习定量模型结构图Figure 3 Structure diagram of deep learning quantitative model
式中:y为肽段的定量值;y′i为预测值。
网络结构参数如表2所示,表示方法与前面的定性模型相同。
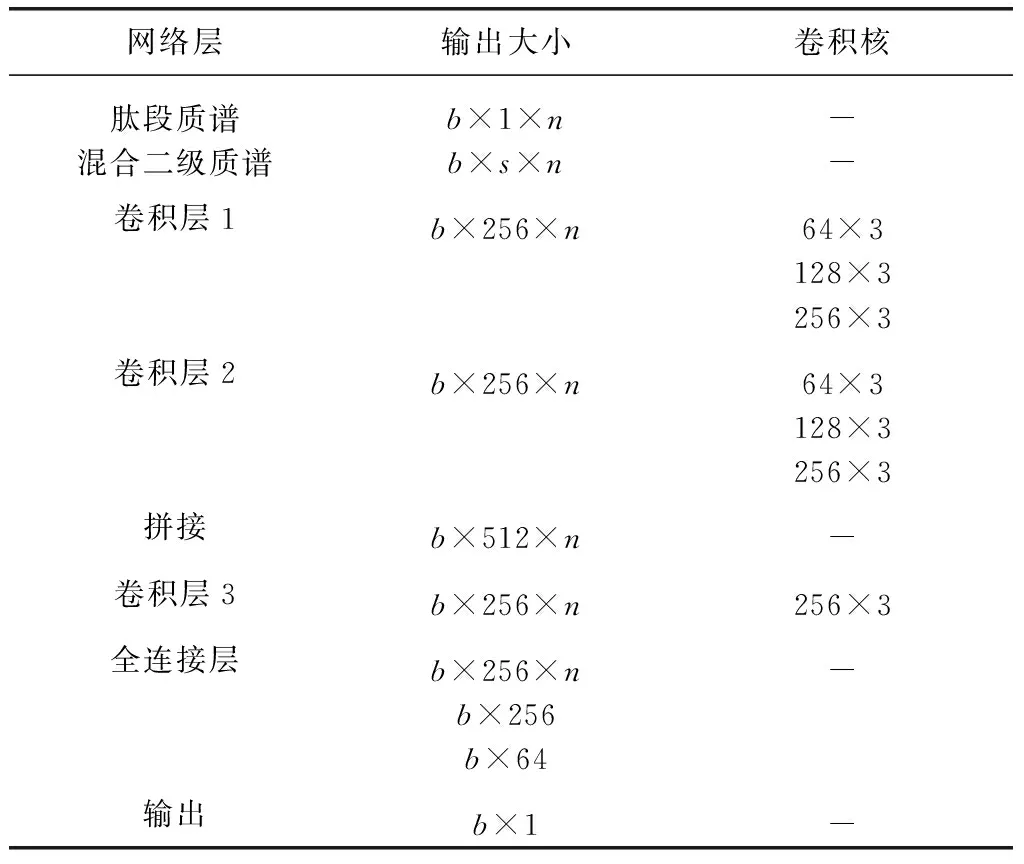
表2 定量模型网络参数Table 2 Network parameters of quantitative model
2 结果
2.1 数据集
在LFQbench[13]论文中提供了很多DIA质谱数据集,是专门用来评估蛋白质组定性和定量准确度而做的实验数据集。质谱数据由3个物种的蛋白质酶解后的肽段,以2种比例分别混合后各进行3次重复SWATH-DIA[14]实验采集得到(人类:[1∶1];酵母:[2∶1];大肠杆菌:[1∶4])。在不同窗口和仪器上进行实验,得到不同的质谱数据。选择其中一个固定窗口的数据作为训练集,另一个可变窗口的数据作为测试集。
为了获取训练数据集的标签,使用蛋白质组定性和定量准确度比较高的方法FIGS[12],在训练集上计算得到定性和定量结果。对于定性模型,将FIGS定性到的肽段作为正样本,将生成的decoy库中的肽段作为负样本(decoy库中的质谱不真实存在,用来混淆target库中的质谱,采用母离子交换-离子峰偏移的方式生成decoy[15])。对于定量模型,将FIGS得到的定量结果,选择比较可靠且准确的定量值作为训练目标。使用LFQbench[13]论文中采用的定量精度评估指标进行过滤,即先使用3次重复实验的定量值计算变异系数(coefficient of variation,cv),再使用cv<0.1过滤,选择A样品和B样品中肽段定量比值比较好的结果。
2.2 定性结果
在肽段定性研究中,为了获取比较可靠的定性肽段,需要同时对decoy库中的肽段进行定性,通过控制错误发现率(false discovery rate,FDR)来获取最终定性到的肽段。使用深度学习定性模型在训练集上优化网络参数后,对测试集进行预测。target和decoy库中的每个肽段都会预测得到一个概率分数,然后计算FDR使其小于0.01。
为了验证模型的准确性,将模型最终定性到的肽段和FIGS定性到的肽段进行对比,如图4所示。可以看到,两种方法定性到的肽段的交集数量为27 788,占深度学习定性肽段的比例为27 788/28 354=98.00%。这说明深度学习模型的定性结果比较可靠。
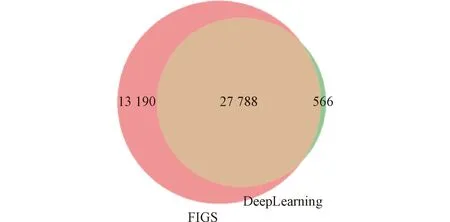
图4 定性结果对比Figure 4 Comparison of qualitative results
同时,统计两种方法在6个样品中均定性到的肽段。FIGS定性交集为18 294个肽段,占总量的比例为18 294/40 978=44.64%;深度学习定性交集为13 680个肽段,占总量的比例为13 680/28 354=48.25%。统计FIGS在6个样品中定性重复率均值为0.578 6;深度学习在6个样品中定性重复率均值为0.662 9。说明深度学习在定性上的重复性很好,因此定性准确度较高。
2.3 定量结果
在肽段定量研究中,为了证明定量方法的准确性和可靠性,通常对重复实验的数据集和不同比例混合肽段进行定量,查看比值结果。目前还没有基于CNN利用DIA色谱信息直接进行肽段定量的研究工作,而FIGS论文使用肽段特有的离子构建色谱峰,该方法的定量准确度很高。所以为了评估深度学习定量模型的效果,使用深度学习模型和FIGS在测试集上分别进行定量。测试集一共有6个文件,包括A样本和B样本的3次重复实验。先获取A样本和B样本3次重复实验均定性到的肽段的定量值,计算cv,保留cv<0.1的肽段,然后取均值作为该肽段的定量值。再计算A样本和B样本中同时出现的肽段的定量值比值,然后与FIGS进行对比,如图5所示。

图5 定量结果对比Figure 5 Comparison of quantitative results
FIGS在其论文中与主流DIA定量软件进行了准确度对比,包括Skyline[16]、OpenSWATH[6]、Spectronaut[17]、DIA-Umpire[18]和 Specter[10]。采用计算绝对中位差(median absolute deviation,MAD)的方式对比准确度,效果明显优于主流软件。本文采用同样的方式与FIGS进行对比,将肽段在B样品中的丰度等分为3部分,计算MAD。如图5(a)所示,本文的深度学习模型与FIGS相比在定量准确度上基本相当。在图5(b)和图5(c)中绘制了肽段在A样品和B样品中的定量值的比值,虚线代表理论比值。从图5(b)和图5(c)中可以看到,深度学习模型定量准确度高的肽段的数量比FIGS明显增多,提高了19.33%[(5 290-4 433)/4 433]。说明与FIGS相比,深度学习能够提高不同丰度下的肽段定量数量。
3 讨论与结论
由于DIA数据是多个肽段同时碎裂产生的混合二级质谱,比较复杂,给肽段定性和定量带来了困难。目前主要基于提取离子色谱图的方法进行定性和定量,但这种方法流程复杂,中间存在误差,色谱图复杂度和色谱时间的不同会导致定性和定量结果不准确。针对该方法存在的问题,本文提出了一种新的肽段定性和定量方法,没有使用色谱维度的信息。利用两个基于CNN的深度学习模型(一个通过二分类的方式进行定性,另一个通过回归预测的方式进行定量),不需要构建色谱峰,也没有提取色谱峰相关的特征,从而减小复杂流程中存在的误差,不受色谱相关因素的影响。本研究在公开数据集上进行了实验,与FIGS对比表明,本文的模型能够提高定性的准确度,经过cv过滤后,绝对中位差指标与FIGS相当的同时,能够显著提高肽段定量的数量,比FIGS提高了约19%,可以有效地对肽段进行定性和定量。
本研究目前在公开数据集上进行了实验,结果表明了方法的有效性,但没有在更广泛的数据集上进行实验,因此本研究存在一定的局限性,需要对模型的泛化能力进一步测试研究。
在未来的工作中,课题组将进一步提高深度学习定性和定量模型的准确度,同时扩展模型的适用性场景,解决更广泛的蛋白质组定性和定量问题。
