Novel miRNA Expression Cassette Downregulates Telomerase Reverse Transcriptase and Inhibits Cell Growth in Retinoblastoma Cells*
2022-12-22ZHAODanDanNGCaiYunWenTao
ZHAO Dan-Dan,DⅠNG Cai-Yun,LⅠWen-Tao
(1)Shanxi Eye Hospital,Affiliate of Shanxi Medical University,Taiyuan 030002,China;2)Central Laboratory,Shanxi Provincial People’s Hospital,Affiliate of Shanxi Medical University,Taiyuan 030001,China;3)Taiyuan University of Science and Technology,Taiyuan 030024,China)
Abstract Objective The present study attempted to design novel miRNA expression cassettes (MEC) targeting specific consensus sequences of hTERT and hTR, which remedied the degradability and cytotoxicity and provided a convenient method for miRNA synthesis.Methods The MECs specific to hTERT and hTR were constructed by overlap polymerase chain reaction(PCR).Telomeric repeat amplification protocol (TRAP)-silver staining and TRAP real-time PCR analysis were used to determine the telomerase activity. The telomere length was determined by real-time PCR, whereas cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The cell apoptosis rate and cell cycle were assessed using the annexin V/propidium iodide (PⅠ) double staining and PⅠsingle staining assays, respectively, coupled with flow cytometry.Results The telomerase-specific MECs were successfully constructed. Each MEC inhibited the telomerase activity differently.Telomerase silencing could induce immediate growth arrest in the G0/G1 phase and led to retinoblastoma (RB) cell apoptosis.Conclusion miRNA-mediated telomerase silencing is an efficient strategy to impair RB cell growth. A robust system must be developed to fully explore the efficacy of miRNAs.The constructed MECs exhibited a strong RNAi effect and thus may be utilized to effectively screen RNAi-targeted sequences for RB gene therapy.
Key words telomerase,miRNA,RNAi,retinoblastoma,gene therapy
Telomere is a special functional complex that comprises tandem repeated DNA sequences and their associated proteins,which are involved in maintaining chromosome homeostasis and integrity[1]. Telomerase is composed of hTR (RNA component) and hTERT(catalytic protein component), which are responsible for telomere maintenance and length regulation[2-3].Telomere maintenance is essential for immortalization of stem cells, cancer cells, and germ cells. Therefore,telomerase activation may be a rate-limiting or critical step in cellular immortality and oncogenesis[4-6].Telomerase prevents degradation by nucleolytic attack, thereby preventing cellular aging and cancer progression. Additionally, it is involved in signaling pathways without apparent involvement in telomere maintenance[7-8]. Most cancers exhibit high telomerase activity, whereas almost all somatic cells exhibit no telomerase activity[9]. Therefore, telomerase is an attractive cancer gene therapeutic target[10].Retinoblastoma (RB) is a common intraocular tumor in children characterized by a protruding mass, which becomes lethal upon metastasis if left untreated.Studies have confirmed telomerase activation in RB cells and contribution of telomerase silencing to RB remission[11].
RNAi mediates knockdown of gene expression in animals and plants, and thus, it has become crucial for functional genomics research in the genome era[12].The mainin vitroRNAi tools include synthetic small interfering RNA (siRNA), short-hairpin RNA(shRNA),and artificial miRNA(amiRNA).Compared with the amiRNA-based design, siRNA and shRNA exhibit disadvantages because of their easy degradability and significant cytotoxicity in host cells.The amiRNA structure is similar to that of endogenous miRNAs, which could avoid the induction of the immune response and reduce offtarget effects by decreasing unwanted passenger strand incorporation into RⅠSC in host cells as compared with shRNA or siRNA constructs[13].miRNA is a 19-24-nucleotide,noncoding,endogenous, single-stranded, and evolutionarily conserved sequence that regulates gene expression post-transcriptionally by binding to complementary sites in the 3' untranslated region (3' UTR) of target genes. amiRNA is either chemically synthesized or designed on a vector such as the plasmid[14].However,the cost of chemical synthesis limits its application in preclinical and clinical trials. Vector-delivered miRNA exhibits long-term safety risks and its clinical application for screening highly effective RNAitargeted sequences is tedious. To overcome the limitation of the chemically synthesized and vectordelivered miRNAs, we designed the miRNA expression cassette (MEC). Overlap polymerase chain reaction (PCR) was used to generate a chimeric DNA template (miRNA) containing H1 promoter that can be delivered into cells directly without cloning into expression vectors. MEC is simple and nontoxic, and thus,it may be a promising tool for genomics research.
1 Materials and methods
1.1 Cell line and plasmids
RB cell (HOX-Rb44) and the plasmid pSUPER retro Neo + GFP with the H1 promoter were obtained from The Cancer Ⅰnstitute of Central South University(Changsha,China).
1.2 Design and synthesis of miRNA expression cassettes
The RNAi target finder tool (http://rnaidesigner.lifetechnologies. com) was used to design three different miRNAs targeting specific consensus sequences, namely MEC-A(whose target was located in the open reading frame (ORF) of hTR mRNA(GeneBank accession NR_001566.1), MEC-B, and MEC-C (whose targets were located in the 3' UTR of hTERT mRNA(GeneBank accession NM_198253).A control miRNA with no significant homology to any known human mRNA was designed.A BLAST search(http://www.ncbi.nlm.nih.gov/BLAST) was used to screen miRNA sequences without any alignment with other sequences in the human genome.The MEC was based upon the rat BⅠC noncoding RNA, with 5'flanking (134-160 nucleotides) and 3' flanking (221-265 nucleotides) sequences.The miR-155 precursor is illustrated in Figure 1a. All primers were designed using Primer 5. The corresponding gene sequences of pri-miRNAs (GMs) were composed of flanking sequences, pre-miRNAs, part of H1 promoter sequences for overlap PCR,and the termination signal of H1 promoter (TTTTT). GMs and all primers were manufactured by Shenggong Company (Shanghai,China). The specific targeting sites and corresponding primer sequences are listed in Table 1. MEC and its synthesis process are illustrated in Figure 1b.

Table 1 Specific targeting site and corresponding primer sequences used in this study
Firstly, PCR1 was performed with Taq Master Mix (Tiangen) and H1F and H1R primers to obtain H1 promoter from pSUPER retro Neo + GFP. The PCR conditions were as follows: 94℃for 3 min; 32 cycles of 95℃for 30 s,55℃for 30 s,72℃for 1 min;followed by incubation at 72° C for 5 min. Then,overlap PCR (PCR2) was used to construct the MEC.The first step comprised the addition of Phusion DNA polymerase(Thermo,USA)and H1F and MR primers for performing 5 cycles of PCR(94℃for 3 min;95℃for 30 s, 50℃for 30 s, 72℃for 1 min). Thereafter,the PCR1 product, GMEC, and Phusion DNA polymerase were added. Then, PCR was continuously performed under the following conditions: 28 cycles of 95℃for 30 s, 66℃for 30 s, 72℃for 1 min;followed by incubation at 72℃for 5 min.

Fig.1 Fundamental structure of MEC and schematic diagram of the overlap extension PCR
1.3 Cell culture and transfection procedures
RB cells were maintained in the logarithmic growth phase at 37℃ in a 5% CO2humidified atmosphere in their own culture medium (RPMⅠ1640 and 20% fetal calf serum). A single MEC and co-transfection groups (MEC-A and MEC-C) were mixed with Lipofectamine-2000TM reagent(Ⅰnvitrogen, Carlsbad, CA), according to the manufacturer's instructions for transfection experiments. Cells were then cultured for different time intervals,harvested,and subsequently analyzed.
1.4 Analysis of MECs transfection and expression
DNA was extracted 48 h after transfection according to the proteinase K and phenol method.The RNA extraction kit (Thermo, USA) was used for total RNA extraction. Quantitative analysis of pre-miRNAs was performed following reverse transcription (RT)reaction by using an RT-PCR kit (Thermo, USA) by SYBR Green mix (TOYOBO, Japan), with GAPDH as the internal reference.The primers for pre-miRNAs:forward,5'-AGGCTTGCTGAAGGCTGTAT-3';reverse, 5'-TGAGTGTACCTTGTTTACCG -3'. For GADPH: forward, 5'-ACCACAGTCCATGCCATCAC-3'; reverse, 5'-TCCACCACCCTGTTGCTGTA-3'.The cycling program was set as follow: 95°C for 3 min;35 cycles of 95°C for 30 s,60°C for 30 s,72°C for 1 min; followed by 72°C for 5 min. The PCR products were detected through 2.5% agarose gel electrophoresis.
1.5 Telomeric repeat amplification protocol(TRAP)assay with silver staining
Telomeric repeat amplification protocol (TRAP)is a classical method for semiquantitative detection of telomerase activity[15]. Culturing was continued for 48 h after transfection, and the protein was extracted according to Kim method. Telomerase activity was determined by TRAP with two primers (TS, 5'-AAT CCGTCGAGCAGAGTT-3' and ACX, 5'-GCGCGG[CTTACC]3CTAACC-3'). Telomerase activity was determined using the TRAP with two primers. The extracted protein, TS primer, and dNTPs were added to each tube at 25° C for 30 min for telomerase binding with GTT sites at TS terminal and synthesizing a 6-base repeat of GGTTAG. Then,telomerase was inactivated at 94°C for 3 min. Taq DNA polymerase and ACX primer were added to tubes for 33 cycles of 95°C for 30 s, 55°C for 30 s,and 72° C for 1 min. Finally, polyacrylamide gel electrophoresis (PAGE) was carried out, using silver staring for developing PCR products and Quantity One software (Bio-Rad, USA) for analyzing the grayscale of each lane on the PAGE gel.
1.6 TRAP with real-time PCR
The PCR-based TRAP assay was used for the absolute quantitative detection of telomerase activity[16]. The primers were the same as those for TRAP assay with silver staining. TSR8 (5'-AATCCGTCGAGCAGAGTTAG [GGTTAG]7-3') is a quantitation standard to estimate the amount of product extended by telomerase in a given extract.The standard TSR8 was five times diluted to eight different concentrations, namely 1, 0.2, 0.04, 0.008,0.001 6, 0.000 32, 0.000 064, and 0.000 0128 mol/L.The PCR conditions were as follows: 30 min at 25°C for telomerase synthesizing a 6-base repeat, 95°C at 10 min for telomerase inactivation and initial DNA denaturation, followed by 40 cycles (at 94°C for 15 s,60°C for 60 s, and elongation at 72°C for 60 s) and a final elongation step at 72°C for 10 min.
1.7 Real-time PCR analysis of telomere length
Telomere length was determined by real-time PCR (RT-PCR)[17-18]. DNA was extracted according to the proteinase K and phenol method at 48 h after transfection.Telomere length was measured by SRBR Green mix (TOYOBO), with 36B4 (encoding acidic ribosomal phosphoprotein) as the internal reference. Telomere primers were as follow: forward(telg),ACACTAAGGTTTGGGTTT GGGTTTGGGTTTGGGTTAGTGT; reverse (telc), TGTTAGGTATCCCTATCCCTA TCCCTATCCCTATCCCTAACA;36B4 primers: forward (36b4u), CAGCAAGTGG GAAGGTGTAATCC; reverse (36b4d), CCCATTCTATCATCAACGGGTACAA. The cycling program for telomere was set as follows: 2 cycles at 95℃for 10 min,95℃for 15 s,and 49℃for 15 s,followed by 40 cycles at 94℃for 15 s,68℃for 15 s,and 72℃for 30 s. The cycling program for 36B4 was as follows:95℃for 10 min;40 cycles of 94℃for 15 s,and 60℃for 40 s. The results were analyzed using the 2-ΔΔCtmethod.
1.8 Cell growth inhibition assay
The 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay was used to detect the inhibition of cell proliferation by MECs in RB cells. Cells in the exponential growth phase were seeded equally onto 96-well plates to acquire a density of 5×104cells/well, and then, the cells were transfected and cultured for 1-5 d. Approximately 20 µl of 5 g/L MTT was added to each well, and the plate was incubated at 37°C with 5% CO2for 4 h.Then, the medium was replaced with 200 µl dimethyl sulfoxide. The absorbance of each well was measured in an enzyme immunoassay analyzer at 490 nm.Every experiment was repeated three times.
1.9 Cell cycle analysis
At 48 h after transfection, the cells were collected and adjusted to a density of 1×109cells/L.Then, the cells were fixed overnight by 75% alcohol.The cells were dyed using 50 mg/L propidium iodide(PⅠ) solution and subsequently analyzed using a fluorescence-activated cell sorting (FACS) scanner(Becton Dickinson, USA) and ModFit software(Verity Software House,USA).
1.10 Cell apoptosis assay
The effect of hTERT knockdown on cancer cell apoptosis was analyzed through flow cytometry. Cells were collected at 48 h after transfection and subjected to annexin V/PⅠdouble staining, according to the manufacturer's instructions (Keygen Biotech, China).The samples were then detected using a fluorescence microscope and a FACS scanner (Becton Dickinson,USA).
1.11 Statistical analysis
All results were analyzed by SPSS software and the average value±standard deviation was used to describe the data characteristics of the results. ThePvalue was used to indicate whether the actual observation results conform to the “invalid hypothesis”.P<0.05 was considered that there was a significant difference.
2 Results
2.1 Construction and validation of MEC
The MECs for each of the targeting sites were successfully established and confirmed through PCR analysis. Agarose gel electrophoresis confirmed the size of single H1 promoters,GMECs,and MECs to be 221 bp, 156 bp, and 365 bp, respectively (Figure 2a).The MEC sequences were obtained by Shenggong Company(Shanghai,China)after gel purification.The secondary structures of artificial pre-miRNAs were predicted using the RNA fold Web Server (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) and were highly similar to those of the endogenic mmu-miR-155 precursor(Figure 2b).

Fig.2 Identification of MECs synthesized by overlap extension PCR
2.2 Validation of MECs transfection and expression
PCR analysis and agarose gel electrophoresis confirmed that the MECs for each of the targeting sites were successfully transfected and expressed.The sizes of pre-miRNAs and GAPDH were 127 bp and 396 bp,respectively(Figure 3).

Fig.3 Identification of MECs transfection and expression by agarose gel electrophoresis
2.3 MECs significantly inhibit telomerase activity
Three MECs specific to hTR (143-164) and hTERT(3 579-3 590 and 3 877-3 897)were MEC-A,MEC-B, and MEC-C, respectively. Telomerase activity was determined by TRAP-silver staining 48 h after transfection. All three target site-specific MECs and the combination group (combination of MEC-A and MEC-C) exhibited significantly higher inhibition rate of telomerase activity than mock and negative control MEC(Figure 4a).RNAi inhibition rates of the MEC-A, MEC-B, MEC-C, and co-transfection groups were (62.39±2.08)%, (68.25±1.34)%, (71.25±1.68)%,and (82.24±2.24)%, respectively, all of which were significantly different from that of the negative control group ((5.60±1.24)%) (P<0.05) (Figure 4b).To further validate the inhibitory effect of MECs on telomerase activity in RB cells, TRAP was applied through real-time PCR to accurately determine the telomerase activity following MEC transfection. Ⅰn the TRAP RT-PCR analysis, telomerase activity was related to the extension products, which were further determined by RT-PCR. Thus, this method directly detected the telomerase activity. The telomerase activity in RB cells was substantially decreased 48 h after transfection. The amounts of telomerase extension products of MEC-A, MEC-B, MEC-C, and co-transfection groups were 32.81±1.53, 23.81±1.56,22.94±2.09, and 14.37±1.23, respectively, which were significantly different from those of the negative control group (62.07±1.82) and mock (64.60±1.56)(P<0.05)(Figure 4c).MEC-B and MEC-C produced a stronger inhibitory effect on telomerase activity than MEC-A, whereas the co-transfection group (MEC-A and MEC-C) exhibited a stronger inhibitory effect than the single MEC group(Figure 4c).

Fig.4 hTERT-specific MECs decreased the telomerase activity in RB cell
2.4 MECs targeting hTERT induced telomere shorten
The telomere length was detected by RT-PCR to determine whether the decreased telomerase activity influences telomere stabilization, which induces cell apoptosis. RT-PCR results (Figure 5) indicated that the telomere length in the MEC-A, MEC-B, MEC-C,and co-transfection (MEC-A and MEC-C) groups was shortened significantly compared with that in the control group NC and mock 48 h after transfection(P<0.05).

Fig.5 Telomerase-specific MECs induced telomere shorten
2.5 MECs targeting telomerase inhibited the proliferation of RB cells
To assess the effect of MECs on cell proliferation, we examined cell proliferation by the MTT assay 48 h after transfection. As shown in Figure 6a, from the second day, the proliferation rates were significantly lower than the control group(P<0.05), and the proliferation inhibition rate of the co-transfection group was higher than that of the single MEC group (P<0.05) (Figure 6a). The proportions of cells in the G0/G1 phase in MEC-A,MEC-B, MEC-C, and co-transfection groups were(49.93±1.19)%, (51.03±0.93)%, (53.78±0.98)%, and(63.21±2.53)%, respectively, which were significantly higher than that in the negative control group((21.40±1.28)% ) (P<0.05) (Figure 6b, c). The findings indicated that the RB cells were arrested at the G1/G0 phase when the telomerase activity was inhibited by MECs.
2.6 MECs targeting telomerase induced apoptosis of RB cells
The RB cells double stained with annexin V/PⅠunder the microscope exhibited green fluorescence in the annexin V-stained membrane and red fluorescence in the PⅠ-stained nucleus.The cell characteristics after annexin V/PⅠdouble staining were annexin V-/PⅠ-for viable cells, annexin V-/PⅠ+for necrotic cells, annexin V+/PⅠ-for early apoptotic cells, and annexin V+/PⅠ+for late apoptotic cells(Figure 7a).A total of 200 cells were collected to determine the apoptosis rates (early apoptosis and late apoptosis) in the MEC-A, MEC-B,MEC-C, and co-transfection groups. The apoptosis rates in the MEC-A, MEC-B, MEC-C, and co-transfection groups were (46.50±2.65)%, (63.00±2.18)% , (66.67±2.25)% , and (79.17±3.06)% ,respectively, which were significantly higher than that in the negative control group ((11.67±1.89)%) (P<0.05) (Figure 7b). Furthermore, the cells stained by annexin V/PⅠwere detected using the FACS scanner(Figure 7c). The apoptosis rates (early apoptosis and late apoptosis) in the MEC-A, MEC-B, MEC-C, and co-transfection groups were (46.47±1.76)%, (55.33±2.93)% , (59.57±3.82)% , and (75.77±3.16)% ,respectively, which were significantly higher than that in the negative control group ((10.03±1.46)% )(P<0.05)(Figure 7d).
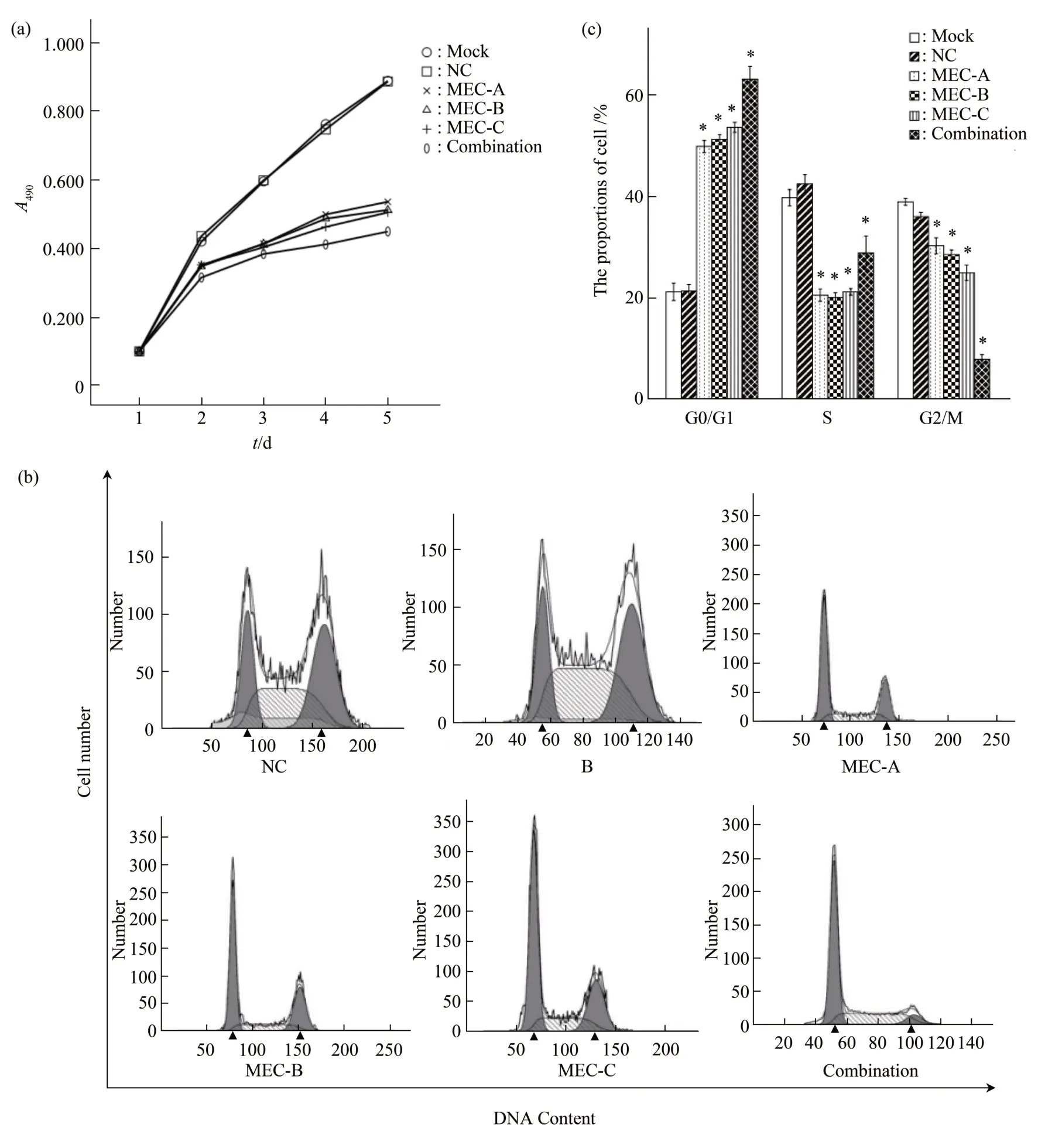
Fig.6 MECs targeting hTERT reduced the proliferation of RB cell

Fig.7 MECs induced RB cells apoptosis
3 Discussion
Telomeres are the protective structures located at the ends of chromosomes, and their shortening decreases the chromosomal stability.Telomerase plays an essential role in the life process of cells by maintaining telomere length. Therefore, telomerase expression defects or overexpression can lead to the occurrence of diseases. Several studies have exhibited that telomerase activation is vital for tumor development and progression[19-21]. The role of telomerase in tumor cells is to prolong telomeres,cooperate with various tumor-related factors,participate in various signaling pathways, and induce tumor occurrence and progression[22-23]. Telomerase participates in the regulation of cell cycle, cell proliferation, cytokine secretion, and tissue differentiation;affects the release of apoptotic cascade signaling molecules; and regulates cytoplasmic calcium ions[24-25]. Therefore, telomerase has become an attractive therapeutic target for cancers.
RNAi technology has provided a new opportunity for the validation of novel therapeutic targets and the development of innovative gene therapies based on the interference with specific gene expression because it efficiently induces sequencespecific gene silencing in mammalian cells[26-27].Studies have exhibited that telomerase is highly activated in RB cell and plays a role in RB genesis and development[28]. Therefore, telomerase expression downregulation by RNAi technology could induce RB cell apoptosis and inhibition of RB cell proliferation.Novel MECs targeting hTERT and hTR were designed to decrease the telomerase activity because siRNA and shRNA could cause significant cytotoxicity. The MECs possessed the human H1 promoter and were manufactured by two-step PCR.MEC can be used for gene therapy because of the lack of cytotoxicity, strong inhibitory effect, and ease of synthesis for RNAi-targeted sequences.
The present study designed three amiRNAs to target different consensus sequences, namely MEC-A(whose target was located in the ORF of TR) and MEC-B and MEC-C (whose targets were located in the 3' UTR). The MECs significantly decreased telomerase activity and accelerated cell apoptosis,which proved that the MECs were an effective tool for producing miRNA and powerful interference.Additionally, the inhibitory effects of MEC-B and MEC-C miRNAs were stronger than that of MEC-A,indicating that the MECs targeting hTERT were effective in eliciting the RNAi pathway in RB cells,which exhibited a strong positional effect. MEC-A and MEC-C were co-transfected into RB cells, and the inhibitory effect of the co-transfected group was stronger than the single MEC effect. The results indicated that combined RNAi was a powerful interference mode that could be applied to the systems with unsatisfactory inhibitory effect of a single MEC.
The impairment of cell growth by specific telomerase targeting can be sustained by the telomere length shortening due to prolonged telomerase activity inhibition or by the loss of telomere lengtheningindependent functions of telomerase[29-30]. Thus, a prolonged decrease in the telomerase activity played a great role in causing the impairment of cell growth as the telomere length was shortened sharply when telomerase expression was downregulated, which also exhibited that the telomere was maintained in a telomerase-dependent manner in RB cells.
hTERT- and hTR-specific miRNAs can efficiently reduce cell growth and induce cell apoptosis by specifically inhibiting the telomerase activity,and miRNA-mediated telomerase suppression represents a powerful tool to counteract RB cell growth. Additionally, the novel MECs developed in this study provide an effective method for screening RNAi-targeted sequences and inducing gene silencing. Moreover, the MEC exhibited particularly simple, high-producing, and low-cost advantages, thus representing a promising technology for genomics research.
