肺泡Ⅱ型上皮细胞在空气污染致特发性肺纤维化中的作用及潜在运用前景
2022-12-17沈洁淼王守林
沈洁淼,王守林
南京医科大学公共卫生学院现代毒理学教育部重点实验室,江苏 南京 211166
空气污染是引发疾病和过早死亡的最重要环境诱因,已成为威胁人类健康的全球公共卫生问题,根据全球疾病负担分析,2019 年空气污染导致667 万人过早死亡[1]。肺部是吸入空气污染物的第一接触点和第一道防线,空气污染物常导致严重的间质性肺病。特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是最常见的间质性肺病,多发生在老年人,其特征表现为肺实质破坏、细胞外基质(extracellular matrix,ECM)沉积及不可逆的肺功能下降,进而导致呼吸衰竭并最终死亡。IPF预后差,诊断后的中位生存期通常为2.5~3.5年[2]。IPF患者伴随着巨大的身体、心理和社会经济负担,且随着全球人口老龄化,IPF 对患者和医疗卫生领域提出了更大的挑战。IPF发病是基因-环境之间复杂相互作用的结果,环境危险因素与IPF 发病密切相关。欧洲的一项回顾性研究发现,长期暴露于空气污染可显著增加IPF 的发病风险[3]。暴露于各种有害刺激引起的肺泡上皮慢性、重复性、亚临床微损伤常导致易感个体随后出现肺泡Ⅱ型上皮细胞(alveolar type Ⅱepithelial cells,AT2)功能障碍,这是IPF致病的启动和核心过程[4]。明确AT2细胞在空气污染致IPF发生发展中的作用和机制,可为空气污染致IPF的潜在干预靶点及效应损伤应对措施提供依据和线索。
1 空气污染与IPF的发病风险
1.1 空气颗粒污染物
空气颗粒污染已是全球疾病负担中重要的环境因素。大气颗粒物水平的增加影响IPF的发病与病程。二氧化氮(nitrogen dioxide,NO2)和臭氧(ozone,O3)是颗粒物的重要成分,主要来源于交通污染,NO2浓度每增加10 μg/m3,IPF 的发病率增加7.93%;尽管目前暂未发现O3与IPF 发病的相关性,但O3可促进IPF 急性加重[5-6]。二氧化硫(sulfur dioxide,SO2)和多环芳烃(polycyclic aromatic hydrocarbons,PAHs)是常见的燃煤污染物,是雾霾发生的重要因素。研究发现,SO2慢性暴露可致小鼠肺纤维化,而苯并[a]芘(benzo[a]pyrene,BaP)和SO2联合暴露可增强SO2促纤维化效应[7],提示空气颗粒物是IPF 的重要触发因素。此外,来自美国和法国的队列研究表明,短期暴露于空气颗粒物与IPF 肺功能加速下降有关[8-9]。
1.2 香烟烟雾
香烟烟雾是室内重要污染物,被公认为是导致IPF 的危险因素[10]。流行病学研究表明,当前和曾经吸烟者发生IPF 的风险高于从不吸烟者,当前吸烟者的风险更高。吸烟与IPF 间存在剂量反应关系,每日吸烟量越大、吸烟时间越长,IPF 的患病风险越高[11]。即使在戒烟多年后,曾经吸烟仍然是偶发性IPF 发展的重要风险因素[12]。与非吸烟者相比,吸烟者会在更年轻时患上IPF,有吸烟史的IPF患者生存期更短[13]。二手烟也是IPF的重要危险因素,可使IPF 风险增加2倍[14]。此外,出生前母亲吸烟的人群患IPF风险会增加1.4倍,这可能与生命早期宫内暴露有关[15]。
1.3 工作场所的空气污染
职业暴露是呼吸系统疾病负担的重要贡献者。美国胸科学会/欧洲呼吸病学会(ATS/ERS)最近的一份声明显示,吸入性工作场所暴露增加了非恶性呼吸系统疾病的负担,其中,IPF的人群归因分值(population attributable fraction,PAF)高于10%(为26%)。对来自11 项研究(总共1 229 例IPF 病例)的PAF分析发现,二氧化硅为3%,木尘为4%,金属粉尘为8%,VGDF(蒸汽、气体、粉尘、烟雾)为26%[16]。一项来自IPF的职业和环境危险因素的病例对照研究发现,可吸入粉尘IPF 风险增加1.4倍,石棉增加1.6倍[14]。
1.4 空气微生物污染
空气微生物特别是病毒吸入,易导致肺纤维化的发生和急性恶化。尽管呼吸道病毒感染与IPF发生之间的直接关系尚未完全确定,但来自先前的严重急性呼吸综合征冠状病毒(severe acute respiratory syndrome coronavirus,SARS-CoV)和中东呼吸综合征冠状病毒(middle east respiratory syndrome coronavirus,MERS-CoV)的证据表明,冠状病毒感染存在异常的肺纤维化后遗症[17-18]。近两年,SARS-CoV-2 在全球大流行,截至2022 年5月,已累计确诊超过5.2 亿COVID-19(corona virus disease 2019)病例。SARS-CoV-2 与先前的SARS-CoV 有79%的序列同源性,因而也可能会出现类似的严重的肺纤维化后果[19-20]。SARS-CoV-2 病毒感染可导致31%的COVID-19 感染者出现急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)[21],而肺纤维化是ARDS 常见的并发症。严重或危重的COVID-19 病例康复患者中,约三分之一的胸部CT 扫描显示肺纤维化病变[22]。对COVID-19住院患者出院2个月后的肺部CT分析,发现约52%的患者显示出广泛的纤维化[23]。由于SARS-CoV-2 流行时间长,波及范围大,感染人数多,因而感染SARS-CoV-2后形成的肺纤维化后遗症,会逐渐成为全球严重的疾病和经济负担,值得广泛关注。此外,最近的一项Meta 分析指出,一些病毒感染可显著增加IPF 的风险(OR:3.48,95%CI:1.61~7.52),包括巨细胞病毒、人类疱疹病毒7和人类疱疹病毒8等[24]。
2 AT2细胞与IPF的发生发展
在正常肺中,肺泡Ⅰ型(alveolar type Ⅰepithelial cell,AT1)细胞覆盖近95%的肺泡表面,主要参与肺泡和毛细血管间的气体交换。肺泡Ⅱ型(AT2)细胞可以分泌表面活性剂,从而降低表面张力以促进呼吸,并以兼性祖细胞的作用而闻名,能够自我更新并在稳态和再生修复过程中产生AT1 细胞[25]。在IPF 肺中,AT2 细胞存在广泛的更新能力受损,吸烟和细颗粒物(particular matter 2.5,PM2.5)污染等引起的衰老、内质网(endoplasmic reticulum,ER)应激、端粒缩短等共同作用,最终导致AT2细胞无法有效修复受损的上皮细胞,由此促进IPF 的发生发展。此外,当AT2 细胞功能障碍时,不能产生足够的表面活性蛋白,肺泡上皮细胞会因机械张力增加而面临继发性损伤,这会加剧上皮细胞耗竭并触发AT2中的促纤维信号,以及抑制AT2细胞分化为AT1,扰乱肺再生过程,形成恶性循环[26]。
2.1 IPF启动期
各种内在(如遗传)和外在(如感染、香烟烟雾和空气污染等)因素刺激,会产生脆弱的AT2 细胞群,脆弱的AT2细胞受到反复的刺激会发展成严重的功能障碍。尽管AT1和AT2细胞均容易受到环境因素作用而损伤,但由于AT2细胞功能障碍使得受损的AT1 难以修复,因而在IPF 中发挥更重要的作用。香烟烟雾或PM2.5里含有复杂的混合物,包括一些重金属和氮氧化物等,易形成自由基,进而攻击AT2 细胞,导致AT2 细胞凋亡或发生ER 应激。此外,吸入的SARS-CoV-2 病毒颗粒首先在鼻黏膜上沉积,随后扩展到肺泡是导致COVID-19 患者病情严重和发生肺纤维化的主要原因[27]。血管紧张素转换酶2(angiotensin converting enzyme 2,ACE2)和跨膜丝氨酸蛋白酶2(transmembrane serine protease 2,TMPRSS2)受体是SARS-CoV-2 的靶点[28],尽管单细胞RNA测序(single cell RNA sequence,scRNAseq)显示只有一小部分AT2 细胞(1%~3%)检测到ACE2,但在肺泡内仍是AT2 细胞主要表达ACE2 和TMPRSS2[29],并且发现AT2 细胞中的ACE2 表达随年龄增长而增加,这可能是老年人特别容易感染SARS-CoV-2 的重要原因[30]。在AT2 细胞内检测到SARS-CoV-2 抗原,证实了AT2 细胞确实可被感染[31]。先前研究报告显示,2003年的SARS-CoV易感染AT2 细胞但不感染AT1 细胞,对AT1 细胞的广泛损害可能是由于感染后的强烈炎症反应所致,而不是直接感染[32],SARS-CoV-2 与之也类似,AT2 细胞似乎优先被感染[33]。根据对SARS-CoV和流感的研究,AT2 细胞可以在病毒感染后启动先天宿主反应,通过分泌多种炎性细胞因子和趋化因子来招募和激活免疫细胞,最终导致功能性表面活性剂丧失、炎症细胞流入以及AT1细胞受损[27]。
2.2 IPF激活期
功能失调的AT2 细胞可引发与免疫细胞,特别是巨噬细胞间的串扰,既可以放大初始损伤事件,又可以促进间充质扩张。肺内巨噬细胞主要分为肺泡巨噬细胞(alveolar macrophages,AMs),包括组织驻留肺泡巨噬细胞(tissue-resident alveolar macrophages,TR-AMs)、单核细胞衍生肺泡巨噬细胞(monocyte-derived alveolar macrophages,Mo-AMs)以及间质巨噬细胞(interstitial macrophages,IMs)。研究发现,Mo-AMs 通常促进伤口修复,在IPF 中持续存在并有助于纤维生态位的形成[34]。失调的肺泡上皮修复与Mo-AMs 激活间存在联系,AT2 细胞分泌的MCP-1 可以促进Mo-AMs 在肺内的募集和激活。体内外模型证明了AT2细胞MCP-1/CCL2分泌在肺纤维化中的重要性,并证实了Mo-AMs 的促纤维化作用。当发生感染或损伤时,单核细胞进入肺泡腔并发育成Mo-AMs,通过基因敲除caspase 8(单核细胞分化为Mo-AMs所必需的基因),发现Mo-AMs而不是TR-AMs 是肺纤维化发展所必需的[35-36]。综上,Mo-AMs被募集到肺中以响应组织损伤,而AT2细胞的损伤促进了Mo-AMs在肺内的募集和激活。
2.3 IPF纤维化期
成纤维细胞是组织衍生的间充质细胞,其核心功能是分泌ECM。在IPF 的发病过程中,活化的成纤维细胞分泌促纤维化介质以增强纤维化环境,导致ECM 的过度产生和向肌成纤维细胞的分化。与成纤维细胞相比,肌成纤维细胞在受损组织中存活时间更长,分泌更多的ECM[37]。目前认可的说法是,反复的AT2细胞损伤引起AT2细胞功能障碍,失调的肺泡生态位持续释放大量促纤维因子,刺激成纤维细胞向肌成纤维细胞分化和ECM沉积,损伤修复异常导致瘢痕形成和肺结构复杂性的进行性丧失,最终导致气体交换功能紊乱和明显的肺纤维化表型[38]。
综上,AT2 细胞功能障碍已成为包括IPF 在内的弥漫性实质肺病的重要过程。AT2细胞作为合成表面活性剂和维持肺泡稳态的肺细胞群,与其他远端肺细胞间存在质量控制网络,以应对异常失调带来的挑战。在外在环境因素反复刺激下,功能失调的AT2 细胞表现出异常的分子特征,包括ER 应激和衰老等,并与免疫细胞和间充质细胞之间异常通讯,最终驱动下游纤维化重塑。
3 AT2 细胞介导的空气污染致肺纤维化过程的主要机制
氧化应激是最常见的促纤维化因素,是IPF 发病机制中的重要途径。香烟烟雾、PM2.5或病毒等损伤AT2 细胞,引起氧化应激增加,刺激活性氧(reactive oxygen species,ROS)产生,进而可能影响各种促纤维化因子如转化生长因子-β(transforming growth factor-β,TGF-β)的激活[10,39]。除了经典的氧化应激机制外,功能障碍的AT2细胞还可通过下列机制发挥促纤维化作用。
3.1 上皮-间质转化(epithelial-mesenchymal transformation,EMT)
EMT是重要的生物学过程,表现为上皮细胞失去细胞-细胞粘附和顶端-基底极性的特征,并获得迁移、侵袭和产生ECM成分的间充质特征[40]。EMT已被公认为IPF 纤维化的关键机制之一。IPF 患者肺组织的免疫荧光显示细胞同时具有上皮和间充质的特征,表明EMT 活跃[41]。与正常AT2 细胞相比,从IPF 患者肺部分离的AT2 细胞表达了更高水平的间充质标志物如α-平滑肌肌动蛋白(alphasmooth muscle actin,α-SMA)[42]。受损的AT2细胞可能是成纤维细胞/肌成纤维细胞的来源,在小鼠肺纤维化模型中的谱系追踪实验表明,表达间充质标志物的细胞具有上皮来源[43]。一些促纤维化因子已被认为是EMT的诱导剂,其中,TGF-β是最相关的因子[44]。当香烟烟雾暴露后,刺激AT2细胞释放TGFβ,进而诱导AT2细胞获得间充质细胞样表型,抑制TGF-β水平后,可减轻EMT 及肺纤维化效应[45]。此外,表观遗传修饰也可影响AT2 细胞的EMT 过程。PM2.5暴露通过甲基转移酶样蛋白3(methyltransferase like protein 3,METTL3)介导的N6-甲基腺嘌呤(N6-methyladenosine,m6A)修饰触发BEAS-2B 细胞的EMT进程并导致小鼠肺纤维化形成[46]。
3.2 细胞衰老
IPF是一种与年龄相关的疾病,85%的患者在首次诊断时年龄>70岁。衰老是肺生理功能下降的主要风险,与年龄相关的衰老失调会导致肺部无法对损伤和压力源做出适当的反应,从而引起细胞损伤修复异常,最终导致死亡,因此衰老与IPF发展的易感性增加有关。除了正常衰老外,吸烟、感染等空气污染会成为“二次打击”源,通过与衰老相关的事件加速肺部衰老,并可能导致AT2细胞再生受损和修复不足,不仅会加速IPF的进展,还会增加老年人本就易受伤害的肺部发生IPF 的风险,并可能成为IPF的最终诱发因素[47]。
AT2 细胞衰老被认为是IPF 的重要病理特征,尽管在间充质细胞中也观察到衰老,但在动物模型中发现是AT2 细胞衰老,而不是间充质细胞的衰老,最终导致肺纤维化的发生[48]。与正常肺组织相比,IPF肺组织AT2细胞衰老相关标志物(如p16)的水平更高。单细胞测序显示,在肺纤维化区域中,AT2细胞衰老相关信号通路异常激活[48-49]。动物及细胞实验也发现,香烟烟雾可以通过SIRT1/自噬依赖性途径诱导AT2细胞衰老,进而诱导肺纤维化发生[50]。衰老的发展和维持涉及一系列复杂信号通路,端粒缩短是引起细胞衰老的原因之一。Povedano等[51]发现敲除AT2 细胞中端粒重复结合因子1(telomeric repeat binding factor 1,TRF1),可引起小鼠肺部端粒功能障碍,并通过诱导DNA损伤和上调p21/p53,引发小鼠肺纤维化。氧化应激和无效的端粒质量控制都可能汇聚导致受损AT2 细胞发生DNA损伤反应。持续的DNA 损伤可激活级联反应导致衰老,包括NF-κB、p53 和PI3K/AKT 通路的激活[2]。衰老的发展伴随着炎性蛋白的分泌,即衰老相关分泌表型(senescence associated secretory phenotype,SASP)。衰老的AT2细胞释放SASP因子,如TGF-β、IL-6等,诱导肺成纤维细胞大量增殖和活化,导致基底膜破坏和ECM的沉积[52]。尽管衰老的成纤维细胞和AT2细胞都可能分泌出SASP,但似乎只有AT2细胞分泌的SASP影响肺纤维化的进展[48],这可能是衰老的AT2 细胞引起IPF 的重要原因,也解释了IPF中炎性基线升高的原因。本课题组近期研究发现,萘[2,1-a]芘(naphtho[2,1-a]pyrene,N21aP,一种六环多环芳烃),可以在AT2 细胞中被细胞色素P450 1A1(CYP1A1)代谢活化生成活性代谢产物N21aP-二氢二醇,形成DNA 加合物,引起AT2 细胞DNA 损伤进而发生细胞衰老,衰老的AT2细胞释放大量SASP 因子,引起AT2 细胞-成纤维细胞间异常通讯,诱导成纤维细胞向肌成纤维细胞分化和ECM沉积,最终导致小鼠肺功能下降和肺纤维化形成。
3.3 ER应激
ER主要参与维持蛋白质稳态,在蛋白质折叠过程中对其进行质量控制。任何影响蛋白质加工的因素都可能使错误折叠的蛋白质在ER 中积聚,称为ER应激。作为对ER应激的反应,细胞会启动一种称为未折叠蛋白反应(unfolded protein reaction,UPR)的信号级联反应[53]。UPR有助于蛋白质合成、折叠和降解,如果UPR不能减轻ER的压力,就会触发凋亡通路[2]。ER 应激和UPR 反应通过AT2 细胞凋亡、EMT 等与IPF 相关[54]。香烟烟雾和颗粒物可引起AT2 细胞氧化损伤和ER 应激,进而导致AT2细胞凋亡[55],这可能是通过p-ERK-eIF2α-ATF4-CHOP 通路介导[56],并在纤维化区域内可见AT2 细胞凋亡标志物表达的增加[57]。AT2细胞产生大量表面活性蛋白,因此对蛋白质稳态特别敏感,表面活性蛋白突变会通过ER应激引起细胞凋亡[2],这也是IPF发生的重要触发因素。
衰老、EMT和ER应激是AT2细胞致IPF的常见机制,虽然这些途径都能单独发挥作用,但更重要的是它们并不是孤立地起作用,而是会相互作用以促进IPF发生发展(图1)。ER应激激活UPR导致促衰老因子的调控,包括p21 和p53,进而引起细胞衰老并释放大量SASP因子。TGF-β是重要的SASP因子,也是EMT 诱导因子,TGF-β在AT2 细胞中引发EMT,由此极化的肺泡上皮细胞获得间充质和迁移特征[58],三者共同作用,促进IPF的发展。
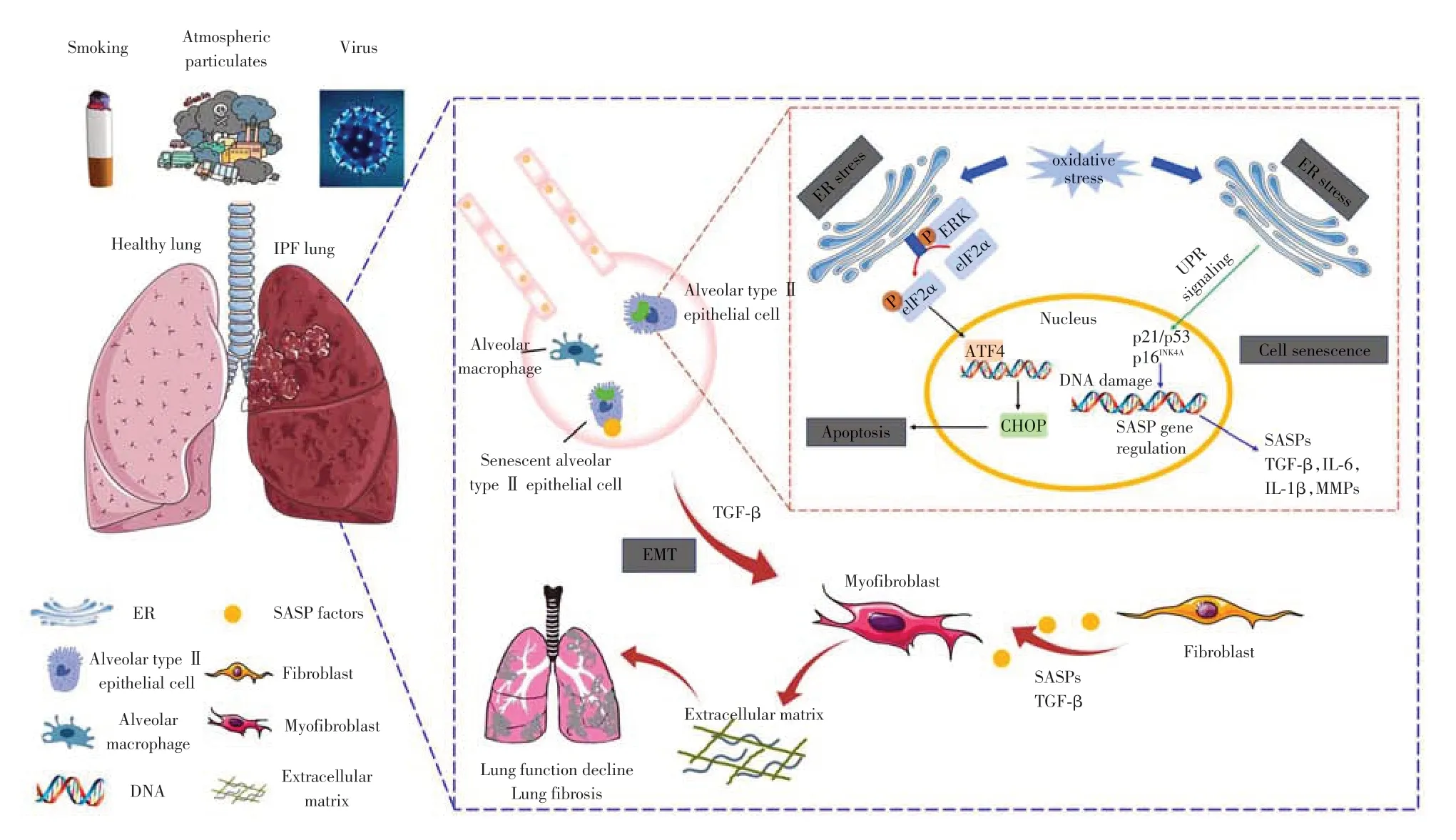
图1 与IPF发生相关的常见生物学过程Figure 1 Common biological processes associated with IPF
4 靶向AT2细胞在IPF预防及治疗中的应用前景
4.1 IPF诊断生物标志物
IPF 的准确诊断仍是一个挑战,肺活检依然是目前诊断IPF的金标准[59],然而由于存在创伤性,通常不会选择。肺部CT 是常用的诊断方法,但有时CT 难以与其他纤维化肺疾病区分。血清生物标志物因其无创性,受到广泛关注,IPF的特定血清生物标志物有助于IPF 鉴别诊断、判断IPF 严重程度、预测对药物治疗的反应以及对临床试验的患者危险程度进行分层以便选择更好的治疗方式。研究表明,可反映AT2 细胞功能障碍的涎液化糖链抗原(krebs von den lungen-6,KL-6)和表面活性蛋白D(surfactant D,SP-D)在诊断IPF 中是有价值的[60-61]。联合使用血清KL-6、SP-D 和骨桥蛋白(osteopontin,OPN)可提高区分IPF患者和非IPF间质性肺病患者的诊断准确性[62]。KL-6 和SP-D 已在日本被用于IPF的诊断[63]。然而,目前IPF血清生物标志物的临床应用仍然有限,仍需多种临床试验验证生物标志物的敏感性和特异性,以期对IPF 进行早期诊断并指导用药,达到最大化的疗效及减少药物不良反应,提供个体化和精准化的治疗。
4.2 IPF靶向治疗和干预
当前,IPF仍是一种不可治愈性肺病,尼达尼布和吡非尼酮是常见的IPF治疗药物,可以减缓IPF的呼吸恶化并延长生存期,但并不能完全改善症状且存在耐受问题。吡非尼酮通过下调关键的TGF-β等的产生和释放,减少氧化应激,尼达尼布是血管内皮生长因子(vascular endothelial growth factor,VEGF)和血小板衍生生长因子受体(platelet-derived growth factor receptors,PDGF-R)的抑制剂,这两种药物均可干预IPF发病有关过程,但均存在一定不良反应,如胃肠不耐受等[64]。重复性AT2细胞损伤是IPF的关键过程,基因和环境之间的复杂相互作用,激活了众多促纤维化途径,它们成为了早期临床试验探索的潜在分子靶点。除了经典的尼达尼布和吡非尼酮外,越来越多的针对潜在分子靶点的新疗法在临床试验中得到探索。AT2细胞衰老是IPF 的关键机制之一,也是IPF潜在的干预靶点,目前首先确定的抗衰老药物是达沙替尼和槲皮素(dasatinib and quercetin,DQ),它们联合使用能在博莱霉素诱导的小鼠肺纤维化模型中有效消除衰老的AT2 细胞并改善肺功能[65]。首次在IPF 患者(n=14)中进行的DQ 临床研究显示,抗衰老疗法是安全的,并且可以改善IPF 患者的身体机能,而相关不良事件如胃肠道不适等可以接受[66]。鉴于IPF肺部的AT2细胞数量较少,消除衰老的AT2细胞可能会阻止肺组织修复,因而必须仔细测试用于治疗IPF的抗衰老疗法,而靶向损伤的AT2 细胞释放的SASP 因子似乎是阻断IPF 有害损伤的另一种可行策略。遗憾的是,尽管已认识到IPF 是由上皮驱动的肺纤维化疾病,但由于机制尚未完全清楚,因此针对AT2细胞的治疗尚未促使患者病程取得实质性进展,未来还需继续努力寻找新的可靠的治疗方法。
5 总结和展望
IPF是一种以ECM过度沉积和肺组织结构重塑为特征的慢性肺病,生存及预后差。越来越多的文献支持空气污染和IPF之间的因果关系。本文阐述了AT2 细胞作为重要的靶损伤细胞在空气污染致IPF 发生发展中的重要作用以及常见的机制,以期为AT2细胞作为空气污染致IPF的潜在干预靶点提供依据。目前,关于IPF的临床诊断仍以肺部CT为主,血清生物标志物因其无创性和特异性,与肺部CT 相比,对IPF 的鉴别诊断及精准治疗更有优势。目前常用的两种药物尼达尼布和吡非尼酮仅能缓解而不能治愈IPF,鉴于AT2细胞在IPF发生发展中的重要作用,尽管目前未取得实质性进展,但相信靶向AT2 细胞的治疗会成为未来IPF 治疗的热点,而深入了解AT2细胞在IPF中的作用机制会对该类药物的研发提供线索和启发。
随着人口老龄化、空气污染的加重及吸烟人数的增多,IPF的患病率整体呈升高趋势。吸烟是IPF最公认的危险因素,控烟及减少烟草使用对IPF 的预防起着重要作用。空气颗粒物及工作场所的风险暴露评估及污染物接触限值的制订,可用于健康保护。疫苗接种可以预防病毒感染,而核酸检测是早期发现SARS-CoV-2 感染的快速方式,这都是目前预防SARS-CoV-2 的有效手段。因此,预防及控制危险因素的暴露,提高高危人群的精准筛查,实行早期诊断和干预,对于预防IPF的发生、减轻居民疾病负担有着重要的公共卫生意义。
