肺动脉高压药物Selexipag的合成工艺研究
2022-12-13邵俊兰范文华焦小雨项良华唐春雷
邵俊兰 范文华 焦小雨 丁 蕾 项良华 唐春雷
(江南大学 生命科学与健康工程学院,江苏 无锡 214000)
肺动脉高压指的是在海平面、静息状态下,经右心导管测量的平均肺动脉压(mPAP)≥3.33 kPa(25 mm Hg),最终可致患者右心功能衰退和死亡。我国肺动脉高压患者的3年生存率为39%,5年生存率仅为21%[1]。因此,抗肺动脉高压药物的研制与工艺改进仍是药物研发工作的热点。
Selexipag(化合物1,结构式见图1,商品名Uptravi,又称赛乐西帕、司来帕格),其化学名称为2-[4-[N-(5,6-二苯基哌嗪-2-基)-N-异丙基氨基]丁氧基]-N-(甲磺酰基)乙酰胺,由瑞士爱可隆(Actelion)生物制药公司和日本新药株式会社联合研发,新药申请于2015年12月21日获美国FDA审评[2]。Selexipag是一种口服有效、高选择性和长效的前列环素(IP)受体激动剂,可松弛血管壁平滑肌,改善损伤的肺动脉内皮依赖性舒张。2015年《欧洲肺动脉高压诊治指南》[3]和2018年《中国肺高血压诊断和治疗指南》[4]皆建议,应使用Selexipag治疗心功能分级为Ⅱ或Ⅲ级的肺动脉高压患者。2021年7月30日,FDA批准Selexipag静脉注射剂(Uptravi IV)用于治疗目前已处于口服疗法阶段,但暂时无法接受口服疗法,WHO功能分级Ⅱ-Ⅲ的肺动脉高压成人患者[5]。2021年12月7日,Selexipag由国家药品监督管理局批准进口注册,用于成人肺动脉高压治疗。
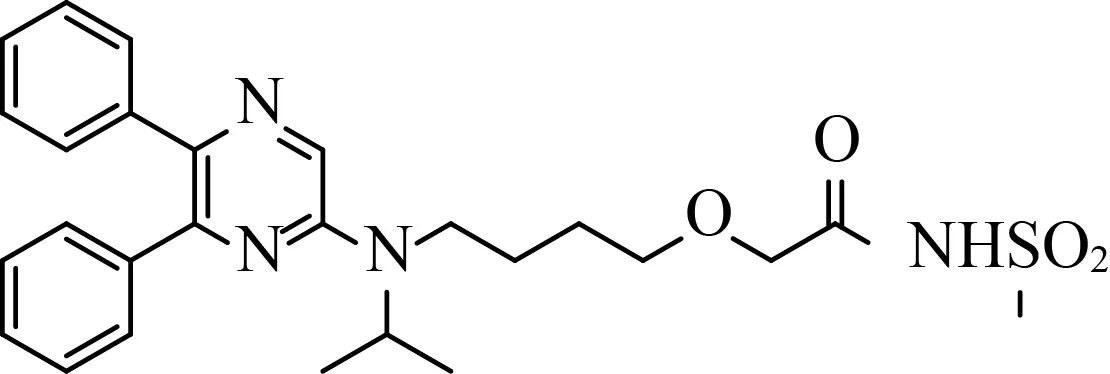
图1 Selexipag的结构
原研公司文献报道了Selexipag的合成路线[6,7],如图2所示。路线1包括以下步骤:(1)以4-氨基-1-丁醇(化合物2)为起始原料,在氧化铂催化下,进行高压还原缩合得到4-异丙基氨基-1-丁醇(化合物3);(2)化合物3和2-氯-5,6-二苯基吡嗪(化合物4)在190 ℃下高温缩合,得到中间体4-(N-(5,6-二苯基吡嗪基)-N-异丙基氨基)-1-丁醇(化合物5);(3)化合物5与溴乙酸叔丁酯发生缩合反应,得到2-(4-(N-(5,6-二苯基吡嗪基)-N-异丙基氨基)正丁氧基)乙酸叔丁酯(化合物6);(4)化合物6水解脱去叔丁酯基,得到 2-(4-(N-(5,6-二苯基吡嗪基)-N-异丙基氨基)正丁氧基)乙酸(化合物7);(5)化合物7先与N,N′-羰基二咪唑(CDI)反应,然后在 1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)存在下与甲基磺酰胺发生酰化反应,得到终产物Selexipag。

图2 Selexipag的合成路线1
原研专利的合成路线步骤为5步反应,该路线存在以下缺点:步骤(1)高压氢化,反应能耗大,工业化需要特殊设备,风险较高;步骤(2)需要190 ℃反应10 h,条件苛刻且存在安全隐患,同时2-氯-5,6-二苯基哌嗪较为昂贵且不易得到,增加了工艺成本,并且由于2-氯-5,6-二苯基哌嗪反应活性较低,此步收率仅有56%;步骤(3)中氢氧化钾固体在苯溶液中难溶,中间体5反应不完全;步骤(5)中缩合剂CDI对湿气非常敏感,遇水易分解,反应对溶剂水分要求高;步骤(1)、(2)、(4)、(5)都需要硅胶柱色谱纯化,成本高,效率低,三废严重。综上,整个合成路线成本高且收率低,总摩尔收率仅为26%,反应操作难度大,安全性上也不利于大规模工业化生产和产业化推广。
鉴于以上路线的缺陷,作者通过对起始原料、合成路线、后处理方法进行改造和优化,确定了较优的合成工艺(图3)。此工艺由4-氯-1-丁醇和异丙胺合成中间体3,以5,6-二苯基-2-羟基吡嗪(化合物8)为原料,通过取代反应得到中间体9。中间体9和中间体3通过缩合反应得到关键中间体5,再通过缩合、水解、酰化三步反应得到目标化合物1。改进后的工艺路线总收率由原来的26.00%提升至40.70%。
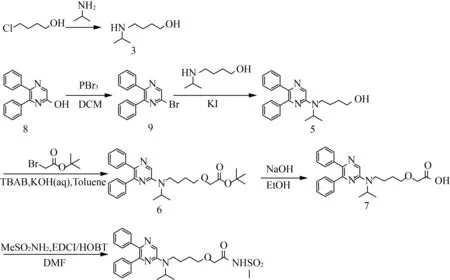
图3 Selexipag的合成路线2
1 实验部分
1.1 主要仪器与试剂
LCMS-8080液相色谱质谱联用仪(日本岛津公司);LC1260 色谱仪(安捷伦科技有限公司);Bruker AVIII-400MHz核磁共振仪(TMS为内标);WRS-1B数字熔点仪(上海仪电物理光学仪器有限公司,温度未经校正);低温恒温搅拌反应浴(郑州科泰实验设备有限公司);集热式恒温加热磁力搅拌器(郑州科泰实验设备有限公司);紫外线分析仪(郑州科泰实验设备有限公司)。
实验所用试剂均为市售分析纯或化学纯,未进一步纯化。
1.2 合成部分
1.2.1 4-(异丙氨基)丁醇(3)的制备
在500 mL反应瓶中加入60.00 g(0.56 mol)4-氯-1-丁醇、131.23 g(2.22 mol)异丙胺,油浴加热至60 ℃反应6 h。高效液相色谱监测反应完全,冷却至室温,向反应液中缓慢加入150 mL浓度为2 mol·L-1的盐酸溶液,调至pH=2,用二氯甲烷(200 mL)洗涤,水相加150 mL浓度2 mol·L-1氢氧化钠溶液调至pH=11,用二氯甲烷(60 mL×3)萃取,合并有机相,无水硫酸钠干燥,抽滤,滤液减压浓缩,得60.58 g黄色透明油状产物4-(异丙氨基)丁醇(3),收率82.50%。ESI-MS m/z:132.1[M+H]+。1H NMR (400 MHz, Chloroform-d)δ:4.14(s,1H),3.43 (t,J=5.4 Hz, 2H), 3.32 (s, 1H),2.67 (p,J=6.3 Hz, 1H), 2.49 (t,J=5.8 Hz, 2H),1.54~1.42 (m, 4H), 0.94 (d,J=6.4 Hz, 6H)。
1.2.2 2-溴-5,6-二苯基吡嗪(9)的制备
在500 mL反应瓶中加入50.00 g(0.20 mol)5,6-二苯基-2-羟基吡嗪(8),溶于300 mL无水二氯甲烷中,室温搅拌下,向其中缓慢加入64.26 g(0.24 mol)三溴化磷,反应5 min后升温至回流,反应1.5 h,薄层色谱监测反应完全(展开剂为石油醚-乙酸乙酯,体积比10∶1,Rf=0.8)。冷却至室温,减压旋蒸除去二氯甲烷,再加入乙酸乙酯(100 mL)、冰水(40 mL),用乙酸乙酯(100 mL×3)萃取,合并有机相,依次用冷却的碳酸氢钠溶液(100 mL)、水(100 mL)、饱和盐水(200 mL×2)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,得到灰色固体即2-溴-5,6-二苯基吡嗪的粗品。然后用石油醚(30 mL)打浆0.5 h,抽滤,滤饼于40 ℃真空干燥箱干燥2 h,得到白色固体2-溴-5,6-二苯基吡嗪(9)59.06 g,收率为95.26%。熔点:138.1~140.7 ℃,ESI-MS m/z:311.0[M+H]+,1H NMR (400 MHz, DMSO-d6)δ:8.91(s, 1H), 7.50~7.23 (m, 10H)。
1.2.3 4-(N-(5,6-二苯基吡嗪-2-基)-N-异丙基氨基)-1-丁醇(5)的制备
向250 mL反应瓶中依次加入58.00 g(0.19 mol)2-溴-5,6-二苯基吡嗪(9)、73.60 g(0.56 mol)4-异丙基氨基-1-丁醇(3)和8.29 g(0.05 mol)碘化钾,加热至120 ℃反应6 h,薄层色谱监测反应结束且副产物较少(展开剂为石油醚-乙酸乙酯,体积比10∶1,Rf=0.8)。将反应液降温至100 ℃,反应液由褐色变黄,加入水(200 mL),用乙酸乙酯(200 mL×2)萃取,合并有机相,无水硫酸钠干燥,抽滤,滤液经减压浓缩得N-(5,6-二苯基吡嗪-2-基)-N-异丙基氨基)-1-丁醇粗品。用石油醚/乙酸乙酯(30 mL,体积比15∶1)混合溶剂打浆,得44.70 g 淡黄色粉末状固体4-(N-(5,6-二苯基吡嗪-2-基)-N-异丙基氨基)-1-丁醇(5),收率65.13%。熔点:101.8~103.2 ℃,ESI-MS m/z:362.2[M+H]+。1H NMR (400 MHz, DMSO-d6)δ:8.14 (s, 1H), 7.38 (dd,J=7.3, 2.4 Hz, 2H), 7.31~7.19 (m, 8H), 4.79 (h,J=6.7 Hz, 1H), 4.49 (t,J=5.1 Hz, 1H), 3.52~3.39 (m, 4H), 1.67 (p,J=8.1, 7.4 Hz, 2H), 1.54 (p,J=6.7 Hz, 2H), 1.23 (d,J=6.7 Hz, 6H)。
1.2.4 2-(4-(N-(5,6-二苯基吡嗪基)-N-异丙基氨基)丁氧基)乙酸叔丁酯(6)的制备
向500 mL反应瓶中依次加入44 g(0.12 mol)中间体5、200 mL甲苯、7.74 g(0.024 mol)四丁基溴化铵和150 mL 40%的氢氧化钾溶液,加毕,搅拌10 min, 继续滴加46.8 g(0.24 mol)溴乙酸叔丁酯,室温反应1 h。薄层色谱监测反应完全(展开剂为石油醚-乙酸乙酯,体积比3∶1,Rf=0.7),旋蒸除去甲苯(可回收),浓缩后的反应液用水(200 mL×2)洗涤,乙酸乙酯萃取(150 mL×3),合并有机相,无水硫酸钠干燥,抽滤,减压浓缩得到52.86 g淡黄色油状产物 2-(4-((5,6-二苯基吡嗪基)(异丙基)氨基)丁氧基)乙酸叔丁酯)(6)粗品,收率91.3%。ESI-MS m/z:476.3[M+H]+。1H NMR (400 MHz, DMSO-d6)δ:8.14 (s, 1H), 7.38~7.35 (m, 2H), 7.29 (d, J=3.3 Hz, 2H), 7.28~7.20 (m, 6H), 3.95 (s, 2H), 3.47 (dt, J=28.3, 7.3 Hz, 5H), 2.00 (d, J=7.8 Hz, 2H), 1.63 (d, J=8.9 Hz, 2H), 1.41 (s, 9H), 1.23 (s, 6H)。无需纯化直接用于下一步反应。
1.2.5 2-(4-((5,6-二苯基吡嗪-2-基)(异丙基)氨基)丁氧基)乙酸(7)的制备
向500 mL反应瓶中依次加入52 g(0.11 mol)中间体6,300 mL乙醇,升温至40℃,开始滴加浓度为2 mol·L-1的 NaOH溶液(200 mL),滴毕,将反应体系升温至回流温度,反应45 min,薄层色谱检测反应完全(展开剂为二氯甲烷-甲醇,体积比15∶1,Rf=0.5),浓缩至无液体滴出,得胶状物14.2 g,将此胶状物溶解在500 mL水中,滴加浓度为2 mol·L-1的稀盐酸至淡黄色固体全部析出,此时pH为5~6,过滤,滤饼于40 ℃真空干燥箱干燥2 h,用150 mL异丙醇重结晶,得42.43 g黄色粉末状产物2-(4-((5,6-二苯基吡嗪-2-基)(异丙基)氨基)丁氧基)乙酸(7),收率92.5%。熔点:112.9~114.2 ℃,ESI-MS m/z:418.2[M-H]+。1H NMR (400 MHz, DMSO-d6)δ:12.78 (s, 1H), 8.14 (d,J=6.4 Hz, 1H), 7.39~7.35 (m, 2H), 7.31~7.24 (m, 6H), 7.22 (d,J=5.3 Hz, 2H), 4.79 (qt,J=8.4, 5.7, 5.2 Hz, 1H), 4.06~4.00 (m, 2H), 3.51 (dt,J=11.8, 6.1 Hz, 2H), 3.44 (dd,J=9.7, 5.6 Hz, 2H), 1.70~1.51 (m, 4H), 1.23 (d,J=6.5 Hz, 6H)。无需纯化直接用于下一步反应。
1.2.6 Selexipag的制备
向500 mL反应瓶中依次加入41.90 g(0.10 mol)中间体7,300 mL N,N-二甲基甲酰胺、24.32 g(0.18 mol)1-羟基苯并三唑(HOBT)、34.51 g(0.18 mol)1-乙基-(3-二甲基氨基丙基)碳化二亚胺盐酸盐(EDCI)和14.25 g(0.15 mol)甲基磺酰胺,开启搅拌,室温反应5 h,薄层色谱监测反应完全(展开剂为二氯甲烷-甲醇,体积比10∶1,Rf=0.8),将反应液浓缩除去部分溶剂,剩余物缓慢加入冰水中,析出固体,过滤,收集滤饼,于40 ℃真空干燥箱干燥2 h,得到Selexipag粗品。用二氯甲烷-正庚烷(400 mL,体积比1∶5)重结晶,加热回流溶解固体,冷凝分出100 mL混合溶剂,溶液仍澄清,降温至10 ℃ 左右有晶体析出,抽滤,滤液重复以上精制步骤两次,合并滤饼得40.42 g白色粉末状固体产物Selexipag,收率81.5%,纯度99.44%[HPLC面积归一化法,色谱柱:Ultimate C18柱(4.6 mm×250 mm,5 μm),流动相A:体积分数0.05%的甲酸水溶液;流动相 B:乙腈;梯度洗脱:0~1 min 90% A,10% B;1~5 min 90% A→50% A,10% B→50% B;5~15 min 50% A,50% B;15~20 min 50% A→10% A,50% B→90% B;20~25 min 10% A,90% B;25~40 min 10% A→90% A;90% B→10% B。检测波长:254 nm;流速:0.5 mL·min-1;柱温:25 ℃]。熔点:138.1~140.7 ℃,ESI-MSm/z:497.3[M+H]+。1H NMR (400 MHz, DMSO-d6)δ:11.78 (s, 1H), 8.17 (s, 1H), 7.43~7.38 (m, 2H), 7.34~7.21 (m, 8H), 4.80 (p,J=6.7 Hz, 1H), 4.11 (s, 2H), 3.57 (t,J=5.9 Hz, 2H), 3.45 (d,J=8.4 Hz, 2H), 3.31 (s, 3H), 1.75~1.63 (m, 4H), 1.24 (d,J=6.7 Hz, 6H)。
2 结果与讨论
2.1 中间体5的工艺条件考察
2.1.1 温度和时间的考察
4-(N-(5,6-二苯基吡嗪-2-基)-N-异丙基氨基)-1-丁醇(5)作为制备Selexipag的重要中间体,其收率和纯度很大程度上影响了终产物的收率和纯度。作者考察了30、 60、90、120、150、180 ℃等温度下的反应情况,结果显示,在低温下反应很慢,通常要反应15 h以上,高温下会生成杂质S,结构见图4,最终确定以120 ℃为最佳反应温度。
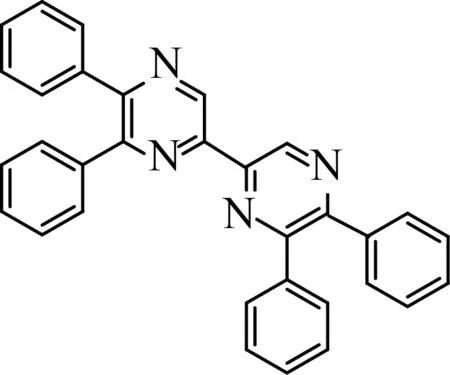
图4 杂质S的结构
作者考察了不同反应时间对收率的影响(图5)。采用HPLC跟踪反应,结果显示,6 h左右反应基本结束,转化率不再随时间延长而增加,经HPLC面积归一法分析其含量也没有太大的变化,优选反应时间6 h 为宜。

图5 反应时间对转化率的影响
2.1.2 对不同缚酸剂的考察
作者考察了缚酸剂对反应收率的影响,结果如表1所示,表明使用碘化钾做缚酸剂收率最高。

表1 不同的缚酸剂对中间体5收率的影响
2.1.3 后处理方法的考察
在对中间体5的后处理过程中发现,中间体5在石油醚-乙酸乙酯(PE-EA)的溶液体系中溶解性不佳,寻找最佳的二元溶剂配比使纯度与收率达到平衡,从而避免繁琐的柱层析操作,不同比例的二元溶剂打浆对中间体5纯度与收率影响如表2所示。
由表2可以看出,当打浆溶剂比例控制在PE∶EA为15∶1时,中间体5经HPLC检测纯度达到99.4%,并且打浆之后的收率也相对其他比例的溶剂更高,因此选择PE∶EA=15∶1作为纯化化合物5的打浆溶剂。

表2 不同比例的二元溶剂打浆对中间体5纯度与收率的影响
2.2 中间体6的制备条件考察
该步反应考察相转移催化剂(实验对照组1~4)和碱(实验对照组1,5~9)对反应收率的影响,相关数据见表3。结果表明,以四丁基溴化铵(TBAB)作相转移催化剂,氢氧化钾(KOH)溶液作碱时,反应收率最高,为91.3%。

表3 合成中间体6的影响因素数据
2.3 化合物1的制备条件考察
考察了影响反应的溶剂(实验对照组1~4)、化合物7与甲基磺酰胺的投料比(实验对照组1,5~11)、缩合剂(实验对照组1,9~11)等因素。控制单一变量进行多次平行实验,实验数据见表4。
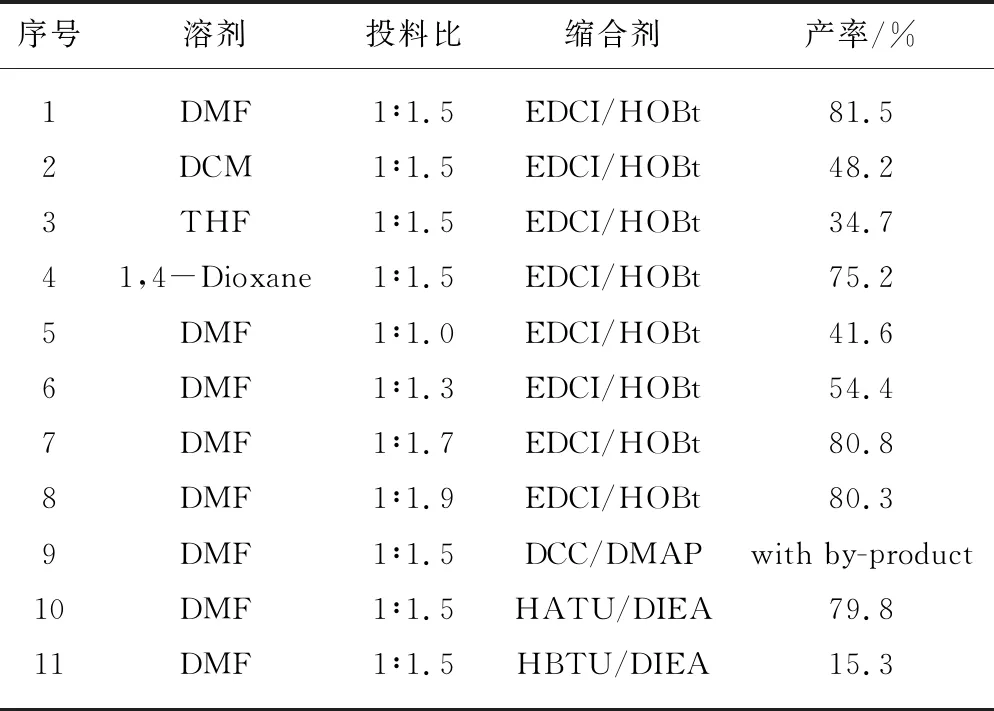
表4 合成化合物1的影响因素数据
确定最优条件为:投料比n化合物7∶n甲基磺酰胺为1∶1.5,缩合剂采用EDCI/HOBt体系,室温反应5 h,收率为81.5%。
3 结论
本研究对Selexipag的各步反应条件进行初步探索,优化了合成方法。第一步中4-(异丙氨基)丁醇(3)的合成与文献报道的高压氢化不同,实验采用无溶剂法反应,后处理简便,反应相对温和,降低反应能耗。中间体5采用价廉易得的5,6-二苯基-2-羟基吡嗪为起始原料经过两步反应得到,收率由原文献的56%提升至67%。中间体6的缩合反应中,引入相转移催化剂TBAB提高反应收率。目标产物Selexipag的后处理无需萃取,加水降温即可析出。作者对关键中间体5、6和目标终产物的制备条件进行了研究,具体包括反应溶剂、时间、温度、投料比、催化剂、缩合剂等因素,反应产物均采用打浆或重结晶的方式纯化,避免柱层析分离,节约成本,便于放大生产,总收率提升至40.70%(以5,6-二苯基-2-羟基吡嗪为基准计算)。研究路线安全环保,简单温和,操作简便,成本低廉,产物收率高,纯度高,具有一定的工业化应用前景,可为Selexipag的生产及其衍生物的合成提供参考。
