Palladium-catalyzed base- and solvent-controlled chemoselective allylation of amino acids with allylic carbonates
2022-12-07YngZhouHngChenPnpnLeiChunmingGuiHifengWngQiongjioYnWeiWngFenerChen
Yng Zhou, Hng Chen, Pnpn Lei, Chunming Gui, Hifeng Wng, Qiongjio Yn,Wei Wng,∗, Fener Chen,c,d,∗
a Pharmaceutical Research Institute, Wuhan Institute of Technology, Wuhan 430205, China
b Chemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, China West Normal University, Nanchong 637009, China
c Engineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai 200433, China
d Shanghai Engineering Center of Industrial Catalysis for Chiral Drugs, Shanghai 200433, China
Keywords:Allylation Amino acid Carbonates Chemoselectivity Palladium
ABSTRACT The utilization of readily available amino acids, which is not only an oxygen nucleophile but also a nitrogen nucleophile, in palladium-catalyzed allylic substitution is realized under mild conditions.The chemoselectivity and multiple allylation are controlled by adjusting the reaction conditions.This represents the first example of this convenient access to valuable N,O-diallylated amino acids.Under the title conditions, a range of amino acids (α-, β-, γ-) and dipeptides can be readily converted in to the corresponding allylic products with excellent yields (67 examples, up to 99% yield) as well as good functional group tolerance.
Amino acids represent a ubiquitous motif found in natural products and pharmaceuticals [1,2].They have been involved in a variety of chemical transformations, especially for the synthesis of artificial amino acids, short peptides and polypeptides with specific biological activities, semi-synthetic antibiotics, new herbicide, and insecticide [3–9].In most cases, these bifunctional substrates have to be protected, either at the amino or at the carboxylic end to ensure selective transformations.For example,amino acid esters have been already widely used inα-alkylation due to theO-protection of the carboxylic group as an easily removable protecting group.In recent years, transition-metal-catalyzed enantioselectiveα-allylation of amino acid ester derivatives has received increasing attention because of the exceptional importance ofα,α-disubstitutedα-amino acids in biological processes; this has been demonstrated by the development of novel nucleophiles (Nunprotected andN,N-disubstituted amino acid esters, aldimine esters, ketimine esters,etc.) [10–15], electrophiles (allylic substrates,vinyl-cyclopropanes, 1,3-dienes,etc.) [16–19], and even highly effi-cient catalytic systems (Scheme 1a) [20–22].However, despite the notable impressive progress achieved in this area ofα-allylation,we were surprised to learn that only few examples have been reported for the asymmetricN-allylation of amino acid esters [23–29].As shown in Scheme 1b, Schmalz and co-works disclosed the stereocontrolled palladium-catalyzedN-allylation of amino acid esters, giving the correspongdingN-allylation products with excellent enantio– and diastereoselectivity [28,29].Although the solubility and selectivity increaseviatheO-protection of the carboxylic group on amino acid, protection may also cause some problems,e.g., increasing the number of synthetic steps and difficulty in deprotecting unstable compounds.Therefore, the development of the direct functionalization of amino acids is greatly required in organic synthesis.
As a matter of fact, compared to amino acid ester as a nucleophile, amino acid contains either N or O reactive sites, which is not only an oxygen nucleophile but also a nitrogen nucleophile(Scheme 1c).Thus, transition-metal-catalyzedN,O-selective allylic substitution of amino acids remains a challenge [30–33].The major challenge inN,O-selective allylation is to control the competition between a nitrogen nucleophile and an oxygen nucleophile on amino acid.What is more, there is still only a very limited number of reports documented in the literature that a carboxy group serves as the nucleophile to afford allyl esters [34–45], probably because of the high reactivity of the resulting allylic esters with metal catalysts.Therefore, we were challenged to develop an effi-cient methodology for such reactions.
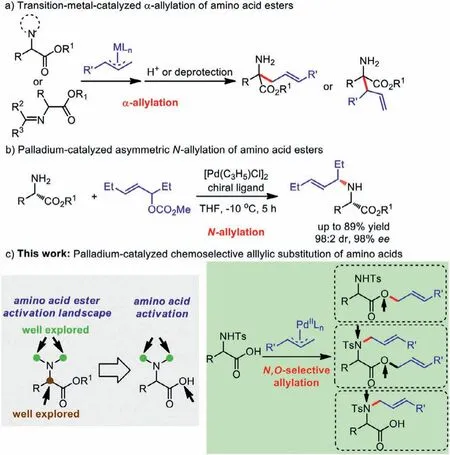
Scheme 1 .Development of allylic substitution of amino acid esters and amino acids.
Herein we report the first palladium-catalyzed chemoselective allylic substitution with amino acids asN- andO-nucleophiles(Scheme 1c).In this context, chemoselectivity for the allylation can be exclusively driven by adjusting the bases and solvents.This new strategy would provide a general and efficient approach to valuable allylated amino acid derivatives.
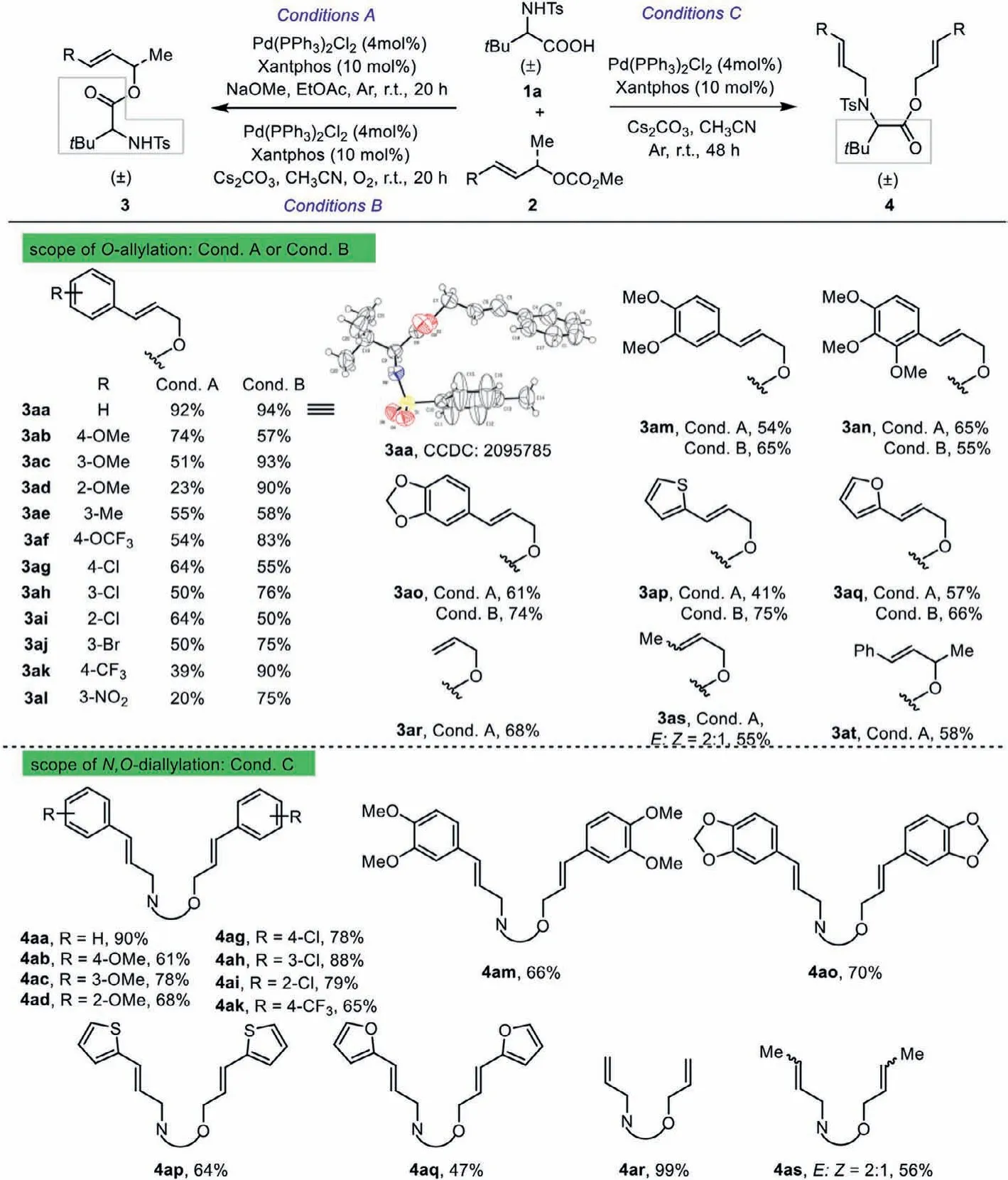
Scheme 2 .Scope of the allylic methyl carbonate coupling partner with racemic tert–butylglycine.For reaction conditions of O-allylation see entries 19 and 15 in Table 1;N,O-diallylation see entry 12 in Table 1.
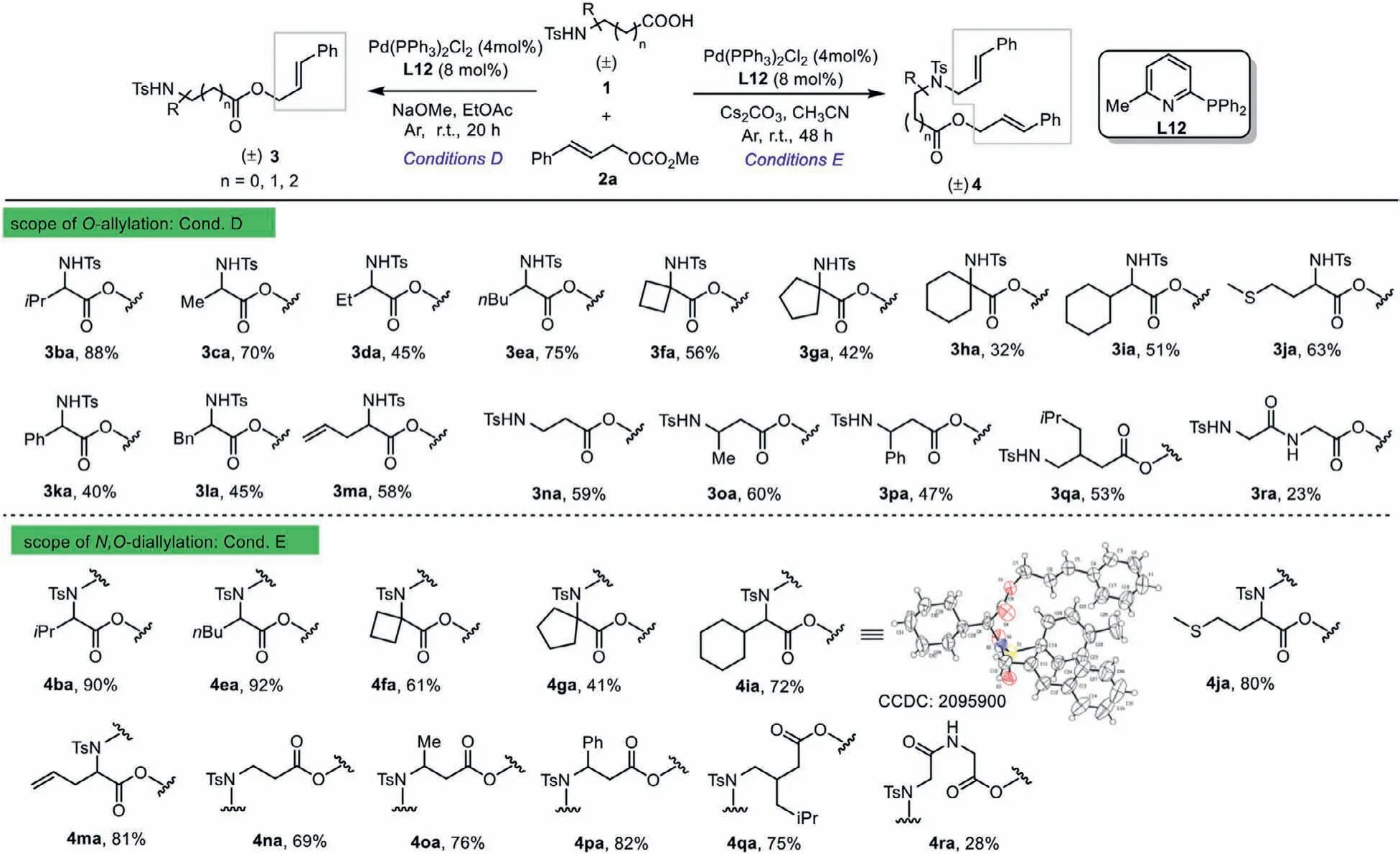
Scheme 3 .Scope of the racemic amino acid coupling partner with cinnamyl methyl carbonate.For reaction conditions of O-allylation see Table S5, entry 11 and N,Odiallylation see Table S5, entry 13.
Our investigation into Pd-catalyzed selective allylic esterifications and aminations began with the evaluation of ligands for coupling racemicN-tosyl–tert-butylglycine 1a and (E)-cinnamyl methyl carbonate 2a.Initially, this reaction was conducted in the presence of 2.5 mol% of Pd(PPh3)2Cl2and 6 mol% of ligand L1 (BINAP).TheN,O-diallylated product 4aa was generated in 35% yield (Table 1,entry 1).To our delight, replacing BINAP with ligand L2 (Xantphos)gave a substantially improved reaction efficiency (Table 1, entry 2,86%).Encouraged by this result, a series of phosphine ligands with different bite angles and electronic natures were screened to identify ligands that would improve the reaction activity and selectivity(Table 1, entries 3 and 4, and Table S1 in Supporting information).As a result, Xantphos still turned out to be far superior.These couplings were all screened using Xantphos as the ligand of choice on palladium, which revealed that only Pd(PPh3)Cl2was suitable(Table 1, entries 5–7 and Table S1).Further screening of the reaction conditions revealed that CH3CN served as the best solvent compared with DCM, 1,4-dioxane, DMF, toluene, EtOAc, THF, DMSO,NMP or CH3OH (Table 1, entries 8–11 and Table S2 in Supporting information).Increasing the amount of catalyst loading and carbonate reagent led to a higher (94%) yield of 4aa (Table 1, entry 12).
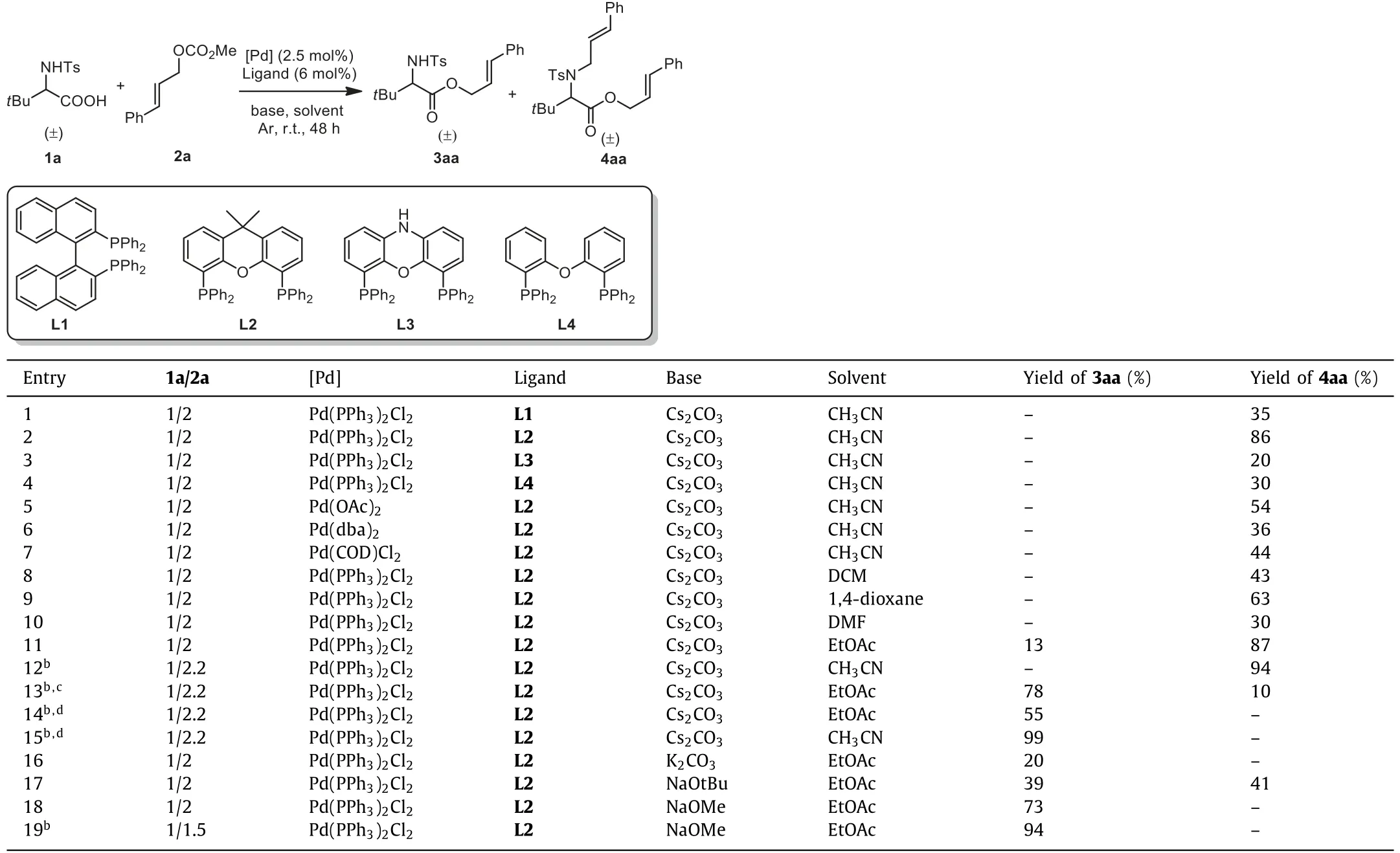
Table 1 Optimization of reaction conditions for racemic N-tosyl–tert-butylglycine and cinnamyl methyl carbonate 2a as substrates.a
Interestingly, this reaction performed in EtOAc gave the allylic esterification product 3aa, though with only a 13% yield,which suggested that the selective allylation ofN-nucleophile orO-nucleophile ontert–butylglycine was possible by controlling the reaction conditions (Table 1, entry 11).Indeed, conducting the reaction under an atmosphere of air or oxygen led to a remarkable increase in yield of the desiredO-allylated product 3aa (Table 1,entries 13 and 14, 78% and 55%).Gratifyingly, using CH3CN as the reaction medium, conversion ofN-Tstert–butylglycine 1a with carbonate 2a under an O2atmosphere resulted in an outstanding yield (entry 15, O2, 3aa, 99%vs.entry 12, Ar, 4aa, 94%).As a Pdcatalyzed allylic C–H acetoxylation [46–48], the beneficial effect of molecular oxygen on theO-allylation reflects its ability to enhance the rate by trapping the putative Pd(0)-alkene species and promote C–O reductive elimination fromπ-allyl-Pd(II) complexes.More importantly, a peroxo-PdIIintermediate could be detected by ESI-HRMS under an atmosphere of ambient O2(Fig.S2 in Supporting information for details), which indicated that molecular oxygen may just kill the catalyst in time to prevent theN-allylation.Finally, a variety of organic and inorganic bases were screened and NaOMe proved to be the best one for theO-allylation product 3aa(Table 1, entries 16–19 and Table S3 in Supporting information).These findings clearly showcase that (a) the allylic substitution favors theO-nucleophile more than theN-nucleophile on amino acids [49] and (b)N-allylic alkylation of amino acids could be suppressed when NaOMe and EtOAc were used as the base and solvent or under an O2atmosphere.When the Ts group was changed to Ac or H, poor results were delivered and only trace amounts of 3aa were obtained.
With the optimal conditions established, we first reacted a representative set of allylic carbonates 2 withN-tosyl–tertbutylglycine 1a to explore the generality of this reaction.It is noteworthy thatO-allylic alkylation oftert–butylglycine was executed under the reaction conditions presented in entry 15 (Conditions B) or 19 (Conditions A) of Table 1.As summarized in Scheme 2,this transformation demonstrated a broad scope with respect to the allylic carbonate reaction partner under both reaction conditions and the correspondingO-allylated products 3aa-3at were obtained with excellent chemoselectivity of up to 94%.The molecular structure of 3aa was unambiguously confirmed by X-ray crystallographic analysis (CCDC: 2095785).An array of cinnamyl carbonates 2b-2k bearing either an electron-donating (e.g., MeO, Me,OCF3) or an electron-withdrawing (e.g., Cl, Br, CF3) group at the phenyl ring were all suitable for the reaction, and afforded the desired allylic esters 3ab-3ak in 58%−93% yields.Notably, themnitro-substituted carbonate was also tolerated and furnished the corresponding product 3al in 75% yield.The presence ofpara(3ab,3af, 3ag, and 3ak),meta(3ac, 3ae, 3ah, 3aj, and 3al), orortho(3ad and 3ai) substitutions on the aryl group proved to be feasible.Furthermore, the substrates 2m-2o with 3,4-di-MeO groups or a bulky fused aryl ring on the phenyl also worked well in this reaction and gave the desired allylic esters 3am-3ao in 65%−74% yields.Importantly, the reactions of 2-thienyl-, 2-furyl-substituted substrates 2p and 2q all proceeded smoothly to provide the products 3ap-3aq in 66% and 75% yields, respectively.Meanwhile, we were pleased to find that our method can also be applied to alkyl carbonates (3ar and 3as, 68% and 55%).Remarkably, 1,3-disubstituted allyl carbonate 2t reacted smoothly toward the correspondingO-allylic product 3at in 58% yield.It is interesting to note that in most cases these two conditions could be complemented each other toward theO-allylic substitution oftert–butylglycine.Subsequently, we focused on the simultaneous construction of C–O and C–N bonds under optimal conditions ofN,O-diallylic substitutions (Table 1, entry 12, Conditions C).The allylation reactions of the carbonates with alkoxy, halogen, trifluoromethyl, and 1,3-benzodioxole groups were successful, producing densely functionalizedN,O-diallylated products 4aa-4ao in 61%–90% yields.What is more, heterocyclic aromatic substrates could also react smoothly withtert–butylglycine 1a to give the expected products 4ap and 4aq in 64% and 47%yields, respectively.In addition, the reaction using 3-alkyl-allyl carbonate provided 4as in moderate yield.
Encouraged by these results, we turned our attention to the investigation of the amino acids scope (Table S5 in Supporting information).Frustratingly, it was found that the optimized protocol of theO-allylation (Table 1, entry 19, Conditions A) proved to be ineffective for theN-Ts valine and a poor yield of the desired allylic ester 3ba was obtained (Table S5, entry 1, 28%).To enhance the activity and selectivity of this reaction, some key factors affecting the performance of this transformation are explored.The best reaction conditions were found with a simple pyridyl-derived phosphine L12 as a ligand when reducing the amount of the base(Table S5, entries 11 and 13).To probe the applicability of the developed protocol, the cinnamyl methyl carbonate 2a was reacted with a set of racemicN-Ts-protectedα-amino acids 1.The results summarized in Scheme 3 show that outstanding chemoselectivities and isolated yields were observed in most cases, independent of the amino acid sidechain.A series of valuableO-allylated andN,O-diallylated amino acid derivatives were finally gained in moderate to good yields (3ba-3ma, 32%−88%; 4ba, 4ea-4ga, 4ia, 4ja,4ma, 41%−92%).The molecular structure ofN,O-diallylated product 4ia was established by X-ray crystallographic analysis.Notably,this transformation was found to be compatible with a series ofβandγ-amino acids with excellent selectivity forO-allylation andN,O-diallylation (3na-3qa, 47%−60%; 4na-4qa, 69%−82%).Besides,dipeptide also gave the corresponding products 3ra and 4ra.
To further demonstrate the potential applications of this protocol, methyl carbonate 2a was reacted with non-racemic amino acids such asN-Ts-D–tert-butylglycine andN-Ts-L-valine, upon which the desired non-racemic allylic amino acid esters (R)−3aa,(R)−4aa, and (S)−3ba were isolated in high yields and without any detectable loss of absolute stereochemical information (Scheme 4a).Furthermore, theN,O-selective allylic reactions could be easily performed on 1 mmol scale (Schemes 4b and c, 72% and 70%).The application of these allylic amino acids generated by this method is demonstrated in Scheme 4.For example, further transformation of 3aa was realized by treatment with I2and NaHCO3in CH3CN, providing the synthetically valuable morpholin-2-one 5 in 54% yield with 1:1 dr (Scheme 4b).Interestingly, productE,E-4aa could undergo an isomerization [50], using our previously reported method of a photocatalyticEtoZisomerization [51] to deliver theZ,Z-isomer in high yield (Scheme 4c,Z,Z-4aa, 95%).More importantly, product 4aa could also be conveniently transformed intoNallylated amino acid 6 with a good yield upon acidic hydrolysis(Scheme 4c, 78%).So far, threeN,O-selective allylation products (Oallylated,N-allylated, andN,O-diallylated) were obtained through the base and solvent-controlledN,O-selective allylation of amino acids.This method provides a new way for the selective modification of amino acids.
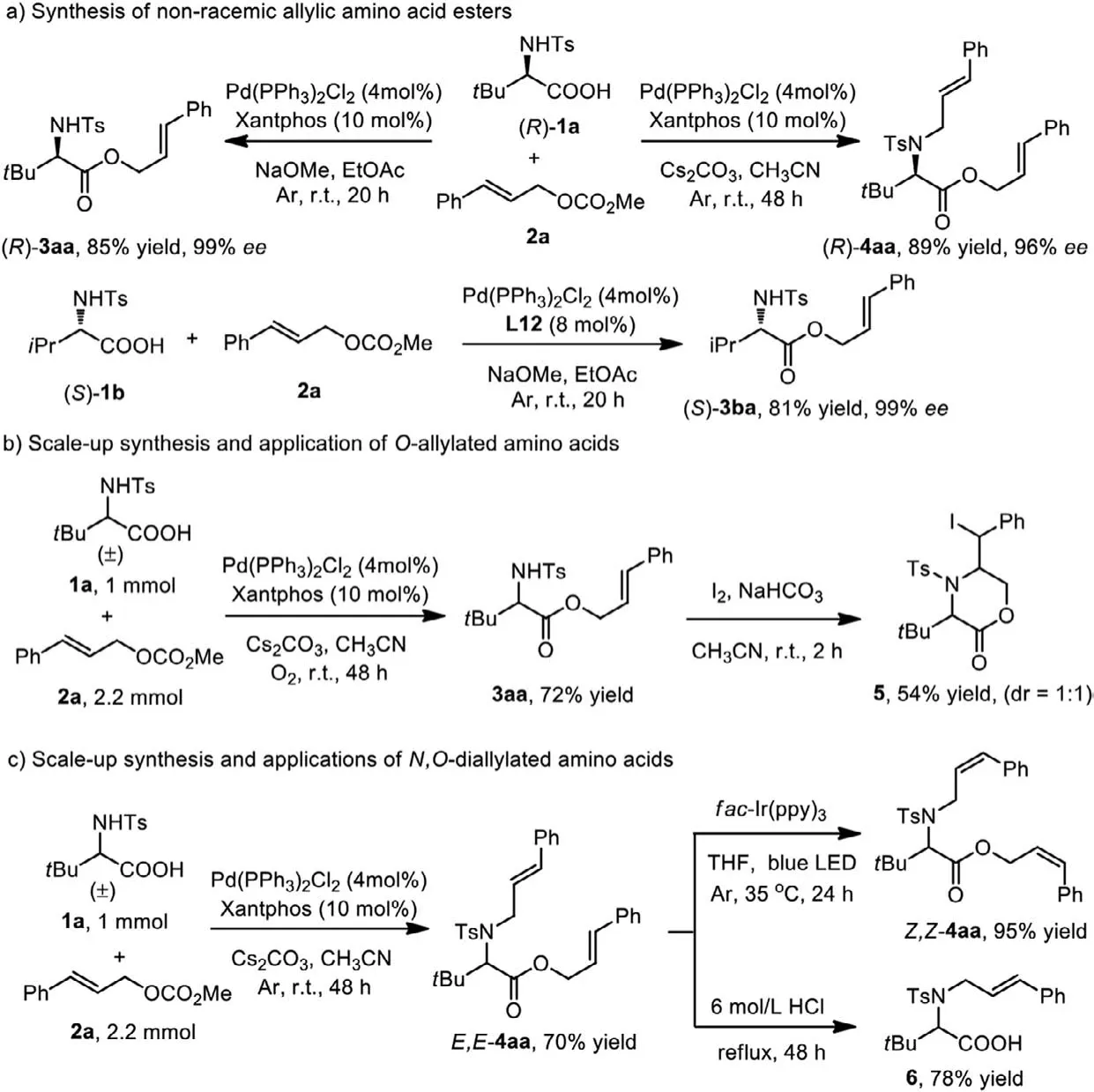
Scheme 4 .Scale-up synthesis and synthetic applications.
To gain insight into the reaction pathway, we examined the reaction processes ofO-allylic andN,O-diallylic substitutions by1H NMR spectroscopy of the crude reaction mixtures (Fig.1 and Fig.S1 in Supporting information).As illustrated in Fig.1, the1HNMR spectra of reaction time clearly show the formation of the desired products 3aa and 4aa under the optimized reaction conditions.The reaction of 1a with 2a gave theO-allylated amino acid 3aa under the conditions A after 5 h, and the1H NMR spectrum showed two doublets atδ6.53,J=15.8 Hz andδ5.18,J=10.8 Hz, and doublet of triplet atδ6.02,J=15.9, 6.7 Hz (Fig.1, I-c).The product 4aa was not observed even extending the allylation time to 48 h.It is important to note that a mixture of 1a and 2a followed by Pdcatalyzed allylation under the conditions C after 1 h afforded 3aa in a good yield, along with a trace amount of 4aa (Fig.1, II-a).As the reaction progress,N,O-diallylated amino acid 4aa content increased, whileO-allylated product 3aa content in the reaction mixture decreased gradually (Fig.1, II-b-d).These results indicates that the transformation of 3aa into 4aa could be crucial for theN,Odiallylic substitution.In addition, as the effect of molecular oxygen,the conversion of 3aa into 4aa was suppressed in the presence of water (Table S4 in Supporting information).
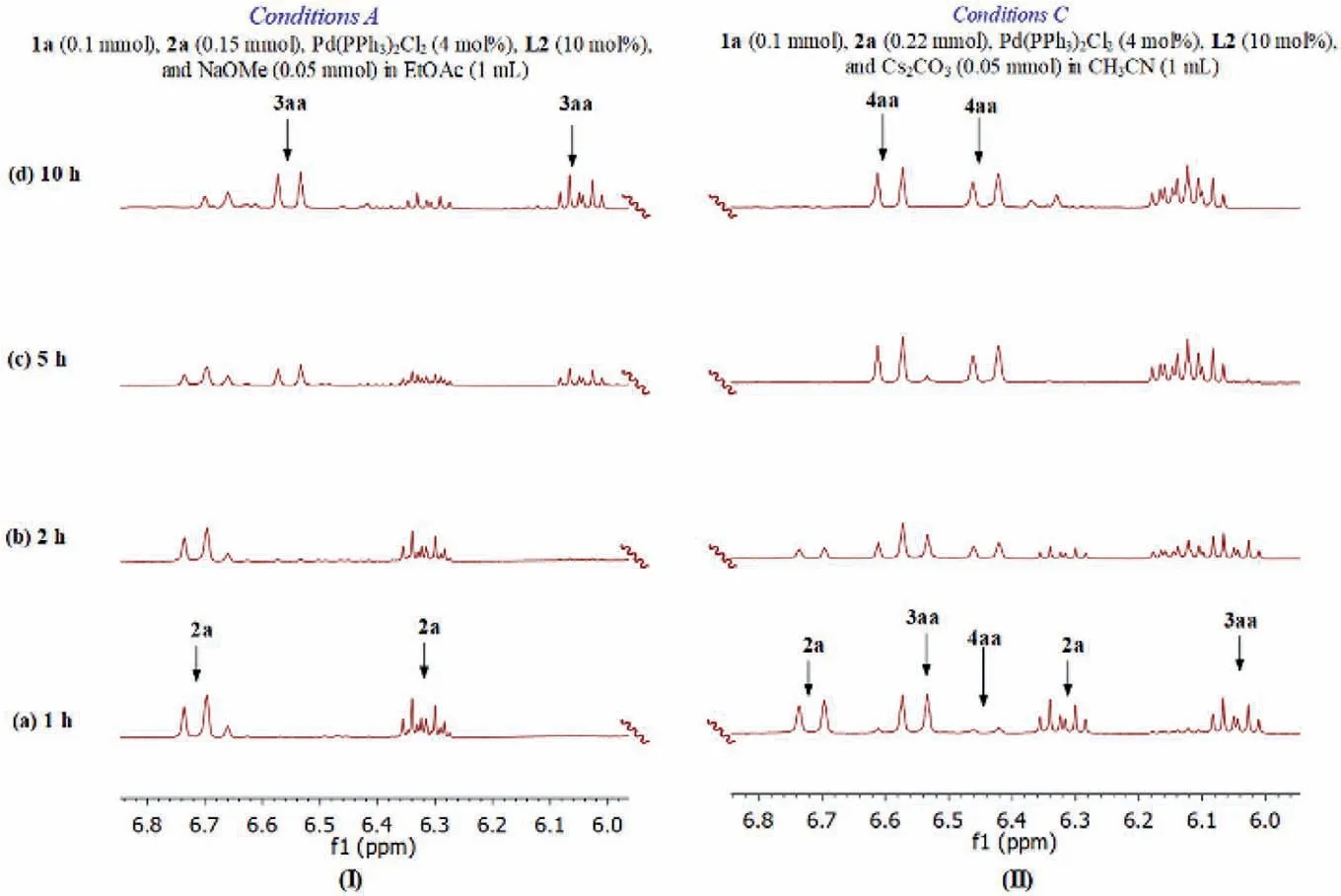
Fig.1 .1H NMR Spectra (CDCl3, 400 MHz); I: performed from 1a (0.1 mmol), 2a (0.15 mmol), Pd(PPh3)2Cl2 (4 mol%), L2 (10 mol%), and NaOMe (0.05 mmol) in EtOAc (1 mL)at room temperature; II: 1a (0.1 mmol), 2a (0.22 mmol), Pd(PPh3)2Cl2 (4 mol%), XantPhos (10 mol%), and Cs2CO3 (0.05 mmol) in CH3CN (1 mL) at room temperature.
On the basis of these results, a plausible mechanism for the Pd-catalyzed chemoselective allylation of amino acids is shown in Scheme 5.Similar to other allylic substitutions, oxidative addition of an allylic carbonate (1a) to Pd(0) gives a Pd-π-allyl species 7.Subsequently, the intermediate 7 underwent esterification with amino acid 1a to form the desired product 3aa and Pd(0).TheO-allylation generated product 3aa which underwentN-allylationviatheN-nucleophilic attack to the intermediate 7, thus forming a new C–N bond and the finalN,O-diallylated product 4aa.When NaOMe and EtOAc are used as the base and solvent, or conducting the reaction under an O2atmosphere or with extra water, the catalytic cycle ofN-allylation is interrupted and the intermediate 9 is not formed during this catalysis.
In conclusion, we have developed the first example of Pdcatalyzed chemoselective allylic alkylation of amino acids with allylic carbonates.The protocol provides a general and efficient approach to various importantO-allylated,N-allylated, andN,Odiallylated amino acid derivatives.The simplicity and availability of the amino acid nucleophile, the mild reaction conditions, and the broad scope of the allylic carbonates are the attributes of the present system.We anticipate additional applications of this strategy for catalytic asymmetric carbon-oxygen and carbon-nitrogen bond formations.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We acknowledge financial support from the National Natural Science Foundation of China (No.21602144), the Science and Technology Program of Sichuan Province (No.2018JY0485), and Scientific Research Project of Education Department of Hubei Province(Nos.Q20211503, B2020057).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.029.
杂志排行

Chinese Chemical Letters的其它文章
- Zeolite-based Fenton-like catalysis for pollutant removal and reclamation from wastewater
- 1,n-Thiosulfonylation using thiosulfonates as dual functional reagents
- Degradation of florfenicol in a flow-through electro-Fenton system enhanced by wood-derived block carbon (WBC) cathode
- Simultaneous determination of indole metabolites of tryptophan in rat feces by chemical labeling assisted liquid chromatography-tandem mass spectrometry
- Self-powered anti-interference photoelectrochemical immunosensor based on Au/ZIS/CIS heterojunction photocathode with zwitterionic peptide anchoring
- The role of Cs dopants for improved activation of molecular oxygen and degradation of tetracycline over carbon nitride