极端中和表型HIV包膜蛋白gp120的高温分子动力学模拟研究
2022-12-07张晓玲杨力权
李 毅, 李 爽, 张晓玲, 曾 新, 桑 鹏, 杨力权
(1.大理大学 数学与计算机学院, 大理 671003; 2.大理大学 农学与生物科学学院, 大理 671003)
1 引 言
获得性免疫缺陷综合症(Acquired Immune Deficiency Syndrome, AIDS),俗称艾滋病,是由人类免疫缺陷病毒(Human Immunodeficiency Virus,HIV)感染引起的烈性传染病. 据世界卫生组织统计,艾滋病在全球广泛传播,严重威胁人类健康. 尽管,终生的抗病毒治疗可将艾滋病缓解为慢性病的范畴、显著延长艾滋病患者的生存时间,但诸如癌症等长期用药的副作用仍然给患者造成巨大的负担.
作为唯一暴露在病毒颗粒表面的病毒编码蛋白,包膜蛋白gp120负责识别和结合宿主细胞表面受体以触发一系列的感染事件[1]. 为使其保守的功能位点免受宿主中和抗体的攻击,gp120利用其序列变异和结构柔性逃避宿主免疫识别[2],进而促使病毒表现出不同的中和表型[3]. 根据多种抗体滴定的实验数据,可将HIV划分为中和抵抗与中和敏感两种极端中和表型[4]. 其中,中和抵抗表型毒株主要分离自HIV长期携带者的血浆,而中和敏感表型毒株则主要来自实验室的传代培养[5].
2015年,研究人员使用氢氘交换耦合质谱的技术观测了多种中和抗体与gp120的相互作用[6]. 实验结果指出,清除病毒效力较高的中和抗体仅在gp120的功能位点引起局部作用,而效力较低的中和抗体则与整个gp120的精细变化有关. 以此进一步推论,由gp120序列变异引起的构象柔性可能是造成HIV不同中和表型的主要分子机制.
因此,我们从gp120结构动态性的角度探索了极端中和表型HIV的分子机制[7]. 从实验标定中和表型的HIV谱系中,随机选择了两株具有极端中和表型的HIV分离株,通过gp120结构模型的构建、分子动力学模拟和自由能图谱分析,初步揭示了HIV中和表型的分子机制. 中和抵抗比中和敏感gp120具有更低的结构稳定性和更高的构象柔性. 病毒包膜蛋白中负责非共价相互作用的结构区域(如layer 1和L1环)在中和敏感gp120中表现出显著更高的构象柔性. 因此,gp120的构象稳定性可能会更易遭到破坏而发生转换. 此外,中和敏感比中和抵抗gp120具有更多涉及协同运动的结构单元. 特别是,在诸如layers 1-2和V1/V2区域的结构单元,运动方向上的差异会使中和敏感gp120更易发生构象转换. 最后,中和敏感比中和抵抗gp120具有更大、更粗糙和更复杂的自由能表面,且中和敏感gp120自由能图谱中的绝大多数自由能最小化区域具有比中和抵抗gp120图谱中更高的自由能水平,表明前者比后者具有更大的构象熵、更丰富的构象状态和更复杂的能力学行为.
尽管HIV中和表型的潜在分子机制已被初步探索,但极端中和表型HIV的热力学分子基础仍待进一步阐明. 一般认为,中和敏感比中和抵抗gp120具有更高的构象柔性以便发生构象转换. 但是,极端中和表型HIV包膜蛋白gp120是否具有不同的热力学性质,其折叠或解折叠是否与中和表型相关等问题仍不清楚. 为此,本文在先前关于极端中和表型HIV毒株包膜蛋白gp120构象柔性差异的研究基础上,在逐渐升高的温度下进行了高温分子动力学模拟,以研究二者在结构稳定性、解折叠和构象柔性上的差异. 研究结果不仅进一步证实了极端中和表型HIV毒株包膜蛋白gp120具有显著不同的构象柔性,还明确了HIV极端中和表型与包膜蛋白gp120热力学性质的关联.
2 材料和方法
2.1结构模型构建
极端中和表型HIV分离株从实验标定的中和表型谱系中随机选取,其氨基酸序列取自蛋白质序列数据库UniProt(http://www.uniprot.org). 中和抵抗毒株H061.14与中和敏感毒株R2的索引号分别为A4ZPW8和Q9WPZ4. 构建gp120结构模型所用模板选自蛋白质结构数据库PDB(http://www.rcsb.org)中冷冻电镜解析的代表性HIV包膜蛋白结构(索引号为5FYJ,分辨率为3.4 Å)[8]. 利用软件包MODELLER V9.15进行同源模建(homology modeling)[9]. 所选模板与靶序列具有高度的序列一致性. 以分子概率密度函数值最小为标准,从构造的20个候选模型中选取具有最佳立体化学质量的结构作为最终模型.
2.2 分子动力学模拟
以上述构建的中和抵抗与中和敏感gp120结构模型为初始,使用GROMACS V 5.1.4软件包[10]和AMBER99SB-ILDN力场[11]进行分子动力学模拟. 将初始结构放置于具有周期边界条件的十二面体盒子中,保证蛋白质中任意原子与盒子边界的距离不小于10 Å. 然后,加入由TIP3P模型[12]描述的水分子,添加Na+、Cl-离子以使蛋白质-水系统在呈电中性的同时还具有150 mM的盐浓度. 为了消除原子间立体化学冲突,并使蛋白质充分浸润于水溶剂中,依次进行了最陡下降法能量最小化和固定蛋白质重原子运动的位置限制性模拟. 最后,为保证模拟系统的稳定性,进行了由1 ns的等容等温(NVT)系综和1.5 ns的等压等温(NPT)系综组成的预模拟.
对于中和抵抗与中和敏感gp120,分别在300 K、373 K和473 K进行了三次30 ns的多复本分子动力学模拟. 每个复本的初始原子速度由相应温度下的麦克斯韦分布随机分配. 具体模拟参数如下:采用LINCS算法[13]约束共价键长,积分时间步长为2 fs;结构快照的输出频率为10 ps;恒压器耦合压强为1 atm,耦合时间常数τp为2 ps;溶质(蛋白质)和溶剂(水分子与抗衡离子)的温度被分别耦合于对应温度下,耦合时间常数τt均为0.1 ps;使用PME算法[14]处理长程静电相互作用,内插阶为4、傅立叶网格为0.16 nm;库伦力的截断半径为1 nm;使用双程截断方案处理范德华相互作用,其中短程和长程的截断半径分别为1nm和1.4 nm;非键相互作用的更新频率设为10个积分步长.
2.3 轨迹分析
轨迹的读取、骨架原子均方根偏差(root mean square deviation, RMSD)的计算,以及解折叠含量Q的测算均使用MDTraj软件包[15]. 使用GROMACS内置的gmx rmsf工具计算残基Cα原子的均方根波动(root mean square fluctuation, RMSF).
3 结 果
3.1 序列和结构比较


图1 极端中和表型HIV包膜蛋白gp120的序列和结构.
3.2 全局结构波动
通过计算轨迹中gp120骨架原子相对于初始结构的RMSD值,可评估模拟过程中gp120的全局结构波动[18]. 从图2可以看出,在所有温度下,尽管中和抵抗与中和敏感gp120在模拟初期具有相似的RMSD值. 但随着模拟的进行,中和敏感比中和抵抗gp120表现出显著更高的RMSD值,说明中和敏感比中和抵抗gp120发生了相对于初始结构更大的构象偏差.
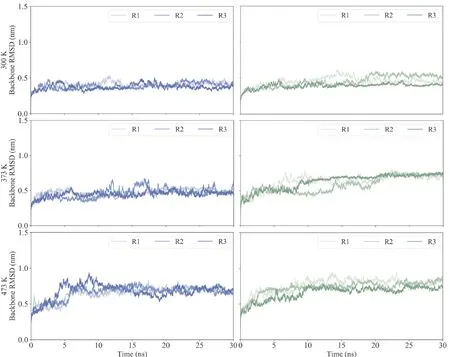
图2 不同温度下,中和抵抗(蓝线)与中和敏感(绿线)gp120三个复本(R1-3)相对于初始结构骨架原子均方根偏差随时间变化的曲线.
在常温下,中和抵抗与中和敏感gp120的RMSD值在0-15ns模拟期间几乎一致. 此后,中和敏感gp120的两个复本表现出比中和抵抗gp120更高的RMSD值,直至模拟结束. 随着模拟温度的升高,中和抵抗与中和敏感gp120的RMSD值均逐渐增大. 尽管如此,中和敏感gp120的RMSD曲线抬升速度要大于中和抵抗gp120,最终导致了前者更大的结构偏差.
以上结果表明,在同一温度下,中和敏感比中和抵抗gp120表现出更大的结构偏差或更高的全局结构波动,说明中和抵抗比中和敏感gp120具有更稳定的结构.
3.3 解折叠程度
为了比较中和抵抗与中和敏感gp120的解折叠程度,不同温度下gp120相对于初始结构的天然接触含量Q[19]随时间的变化显示在图3中. 在常温下,中和抵抗与中和敏感gp120均含有90%左右的天然接触含量. 中和抵抗gp120三个复本的天然接触含量分布较为集中,而中和敏感gp120各个复本则分布在92%至88%之间的区域中,总体上较中和抵抗gp120具有更多的天然接触含量.
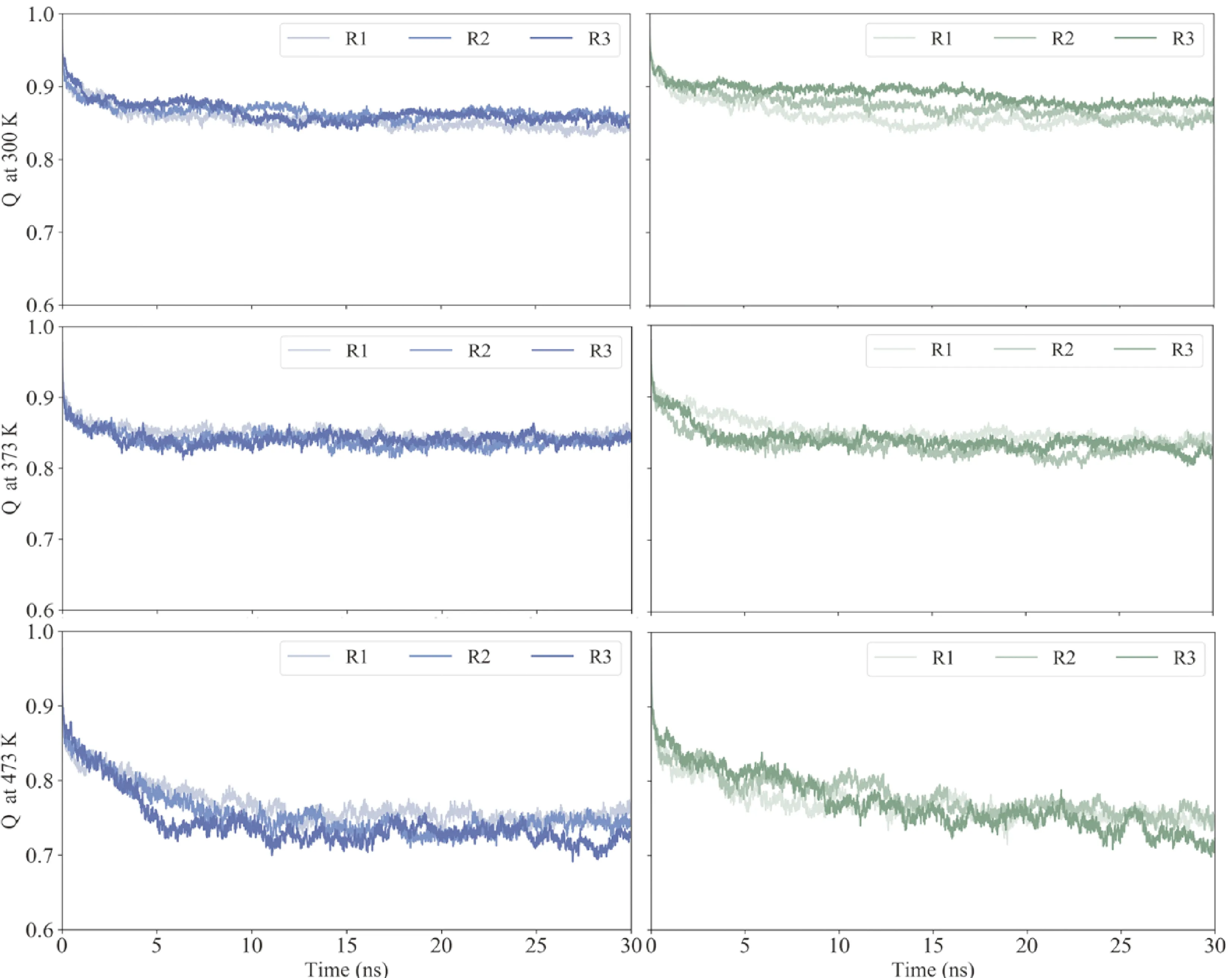
图3 不同温度下,中和抵抗(蓝线)与中和敏感(绿线)gp120三个复本(R1-3)天然接触含量Q随时间变化的曲线.

图4 中和抵抗(蓝色)与中和敏感(绿色)gp120在RMSD和Q空间中的群体分布.
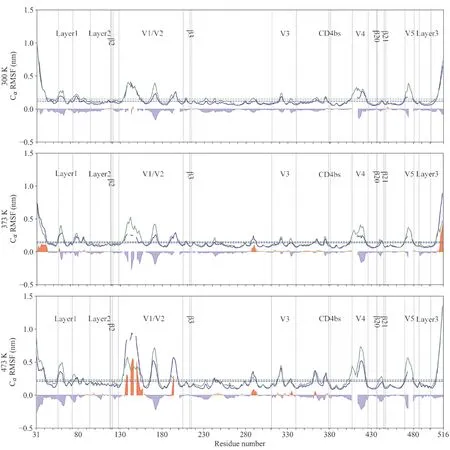
图5 不同温度下,中和抵抗(蓝线)与中和敏感(绿线)gp120的Cα原子均方根波动曲线.
随着温度的升高,中和敏感和中和抵抗gp120的天然接触含量均逐渐降低,并在473 K下低至70%左右. 在373 K温度下,中和抵抗与中和敏感gp120具有非常相似的天然接触含量曲线,说明中和抵抗与中和敏感gp120在该温度下具有相似的解折叠程度. 在473 K模拟温度下,中和抵抗gp120能较快到达较低的天然接触水平,说明中和抵抗gp120的解折叠程度较高.
比较中和抵抗与中和敏感gp120之间天然接触含量的差异可以看出,中和抵抗gp120的天然接触含量总比中和敏感gp120的稍低. 但两者差异较小,说明相对于初始结构的天然接触含量在中和抵抗与中和敏感gp120之间不具备显著差异,也就是中和抵抗与中和敏感gp120具有相似的解折叠程度.
3.4 构象群体分布
为了比较中和抵抗与中和敏感gp120在构象群体分布上的差异,各个温度下的轨迹被投射到由RMSD和Q组成的空间中. 如图4所示,中和抵抗与中和敏感gp120具有相似的构象群体分布范围,均位于RMSD从0.2至1 nm和Q从95%至70%之间的区域. 但是,中和抵抗与中和敏感gp120具有不同的构象群体分布形态. 对于中和抵抗gp120,其构象群体可被划分为两个主要区域,即RMSD约0.3 nm、Q约85%的区域和RMSD约0.8 nm、Q约75%的区域. 而中和敏感gp120则至少具有四个主要的构象群体,即RMSD约0.2 nm、Q约88%的区域;RMSD约0.4 nm、Q约83%的区域;RMSD约0.8 nm、Q约83%的区域和RMSD约0.8 nm、Q约75%的区域.
上述结果说明,中和敏感比中和抵抗gp120在模拟过程中经历了更大的构象变化,因而具有更高的构象熵. 中和敏感比中和抵抗gp120具有更多样的构象群体分布,即具有更丰富的构象多样性.
3.5 局部构象柔性
通过计算中和抵抗与中和敏感gp120每个残基Cα原子的均方根波动RMSF值,可定量描述蛋白质局部结构区域的波动[20]. 此外,中和抵抗与中和敏感gp120的RMSF平均值,以及等同残基位置上中和抵抗gp120的RMSF值减去中和敏感gp120的RMSF值所得到的差值,能进一步反应各个温度下中和抵抗与中和敏感gp120的局部构象柔性.
在常温下,中和抵抗与中和敏感gp120的RMSF平均值分别为0.12和0.15 nm,表明中和敏感gp120具有更高的构象柔性. 除少数区域外,中和抵抗与中和敏感gp120的绝大部分结构区域均具有相似RMSF曲线,说明两者具有相似的构象柔性分布特征. 两者构象柔性差异明显的区域主要集中在V1/V2区域和V4-5环.
随着温度的升高,中和抵抗与中和敏感gp120的RMSF值均随之增大,说明全局构象柔性也相应增大. 但是,中和敏感总比中和抵抗gp120具有更大的RMSF平均值,中和抵抗与中和敏感gp120在373 K下的RMSF均值分别为0.13和0.15 nm,在473 K下的RMSF均值分别为0.21和0.23 nm. 对于在常温下就比中和抵抗gp120较高的区域,V1/V2区域在373 K下仍然维持中和敏感高于中和抵抗gp120的趋势,但在473 K翻转;V4-5环则在两个高温下均呈现出中和敏感gp120较高的RMSF值.
4 讨 论
作为唯一暴露在外的病毒编码蛋白,包膜蛋白gp120在HIV病毒侵染和免疫逃逸中扮演着关键角色[21]. 实验[5]和理论[7]研究都指出其与抗体的相互作用在很大程度上决定了HIV的中和表型. 在我们前期的研究中,极端中和表型HIV的包膜蛋白gp120的结构模型呈现出高度的相似性[7]. 经过分子动力学模拟后,极端中和表型在结构动态性、构象柔性、大尺度分子运动和自由能水平上均表现出差异[22]. 该研究从包膜蛋白gp120动态性的角度初步揭示了HIV中和表型的分子机制.
在本文中,我们进一步研究了包膜蛋白gp120热力学性质与HIV中和表型的关系. 在高温分子动力学模拟中,包膜蛋白gp120表现出与HIV极端中和表型呈正相关的热力学性质,即中和敏感gp120在各温度下均相较中和抵抗gp120具有更大的全局结构波动、更多样的构象群体分布和更高的局部构象柔性. 但是,从天然接触含量可以看出,中和抵抗和中和敏感gp120在各温度下均具有相似的解折叠程度,说明HIV中和表型与其包膜蛋白gp120的解折叠程度无关.
与球状蛋白的解折叠[23]不同,gp120由于除核心结构外,还具有伸展在外的可变环区. 其构象柔性和解折叠程度并不像球状蛋白那样呈现出严格的正相关关系. 而且,gp120高度变异的序列很可能不存在诸如盐桥[24]等强相互作用,以维持其热力学性质. 蛋白质的解折叠可以被记作其折叠成天然构象的逆过程,本研究从高温解折叠热力学的角度明确了极端中和表型HIV背后的分子基础.
5 结 论
在本研究中,延续我们前期关于极端中和表型HIV包膜蛋白gp120的分子动力学模拟,以中和抵抗H061.14与中和敏感R2两个极端中和表型HIV的gp120结构模型为研究对象,通过比较中和抵抗和中和敏感gp120在全局结构波动、天然接触含量、构象群体分布、局部构象柔性等方面的差异,以期阐释HIV中和表型与其包膜蛋白gp120热力学性质之间的关系. 主要结果为:(1)HIV中和表型与gp120的热力学性质呈正相关;(2)中和敏感比中和抵抗gp120表现出更大的结构波动、更多的构象状态和更高的全局构象柔性;(3)二者具有相似的天然接触含量温度,说明HIV中和表型与gp120解折叠程度无关. 总之,本文的研究结果从热力学的角度阐释了HIV包膜蛋白gp120的“结构-热力学性质-表型”三者间的关系,明确了HIV中和表型背后的分子基础.
