添加P2O5对生物活性玻璃力学性能与生物活性的影响*
2022-11-27王宏远王建春杨树青苏欣
王宏远 王建春 杨树青 苏欣
龋病、牙周病及外伤等因素所导致的牙缺失患者数目巨大,牙齿拔除后,因束状骨快速吸收及上颌窦气化效应,失牙12个月后后牙牙槽嵴可水平向吸收50%,上颌前部吸收70%,上颌后牙区骨丧失更可高达80%[1]。伴随着人民生活水平及保健意识的不断提高,人们更倾向于选择种植义齿[2]。骨移植是治疗骨缺损的重要手段,人工合成的骨替代材料(alloplast)拥有诸多优点,能为成骨细胞(osteo blast,OB)提供可以依附的稳定三维空间,分解后的产物更利于成骨细胞的活性,为新骨的形成创造理想的机体微环境,因此越来越多的专家学者致力于开发此类材料[3]。生物活性玻璃(bioactive glass,BG)植入人体后形成的羟基磷灰石通过吸附胶原和细胞与骨组织间生成化学键结合,可有效防止该界面因受到应力出现微裂隙而导致纤维结缔组织长入,影响引导骨再生的效果[2]。有研究发现,生物活性玻璃具有独特的骨诱导能力,其降解产物能够上调宿主成骨基因的表达,引导骨生成[4,5]。为探究性能更优异的生物活性玻璃材料,陈露等人将不同量的磷掺入其中,实验发现研究中制备的生物活性玻璃呈现磷含量越高,新骨矿化生成越多,速度越快的趋势[6]。Tilocca的研究表明,含磷的生物活性玻璃与钠钙硅酸盐玻璃相比,表面呈现出更明显的亲水性,更利降解[7]。由此可见掺入磷元素对生物活性玻璃性能改变有明显影响。本实验为研制性能更优异的成骨材料,以氮氧生物活性玻璃为基础[8,9],用不同含量P2O5取代部分SiO2,并对其力学性能和生物活性进行测定及分析研究如下。
1.材料和方法
1.1 实验材料和设备 本实验过程中所需要的材料和设备如表1、表2所示:

表1 实验材料

表2 实验设备
1.2 研究方法 以氮氧生物活性玻璃(SiO2-CaO-ZnO-Na2O-Si3N4)为基础,制备不同磷含量的基础玻璃,磷源选用P2O5(详见表3)。试件制备采用有机泡沫浸渍工艺将基础玻璃制成多孔生物活性玻璃支架,模板选择高温环境下可完全挥发的PU 海绵,并研究不同含量P2O5对多孔生物活性玻璃支架的抗压强度、抗弯强度、降解性能和体外矿化活性的影响。

表3 A、B、C、D、E、F六组氮氧生物活性玻璃成分百分比(wt%)
1.3 实验过程
1.3.1 基础玻璃的制备 基础玻璃采取高温熔融法制备:按比例称量所需原料,放入陶瓷研钵内研磨,直至混合均匀且手捻无颗粒感,移入氧化铝坩埚内,按设定程序(自室温升至1500℃、保温2 h)于高温箱式电阻炉中烧结,后迅速将熔融态玻璃水淬、烘干,制得基础玻璃(见图1)。

图1 基础玻璃试样
1.3.2 多孔生物活性玻璃支架烧成温度的确定 多孔生物活性玻璃支架的烧成温度应该在基础玻璃的软化温度(softening temperature,Ts)与析晶起始温度(crystallization temperature,Tc)的范围内选取,温度参数以综合热分析测试(differential scanning calorimetry,DSC)获得,测试仪器使用耐驰449F-1,测试气氛Ar 气,升温速率10℃/min,从室温升温至1200℃。得如下结果(图2,表4)。
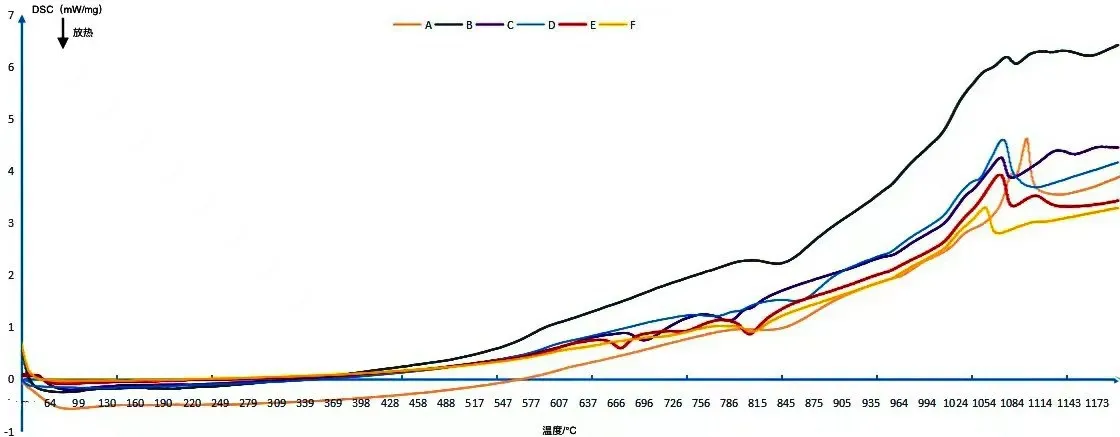
图2 基础玻璃DSC曲线图
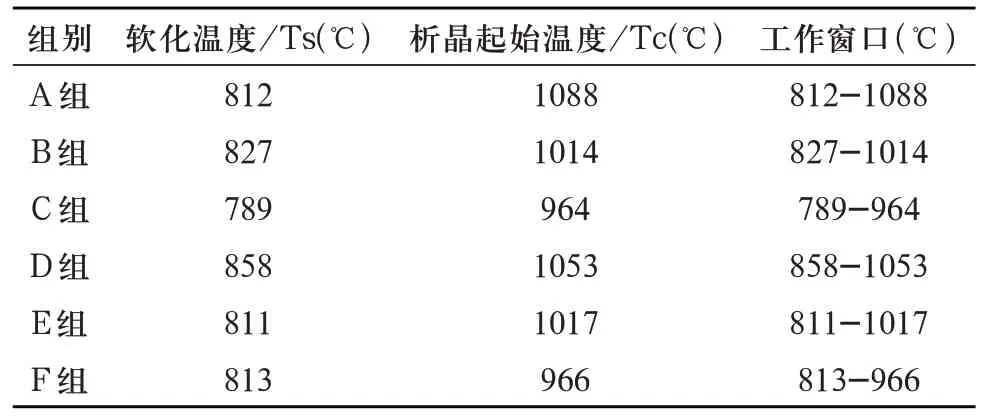
表4 基础玻璃温度参数
1.3.3 多孔生物活性玻璃支架试件的制备 多孔生物活性玻璃支架试件制备采用有机泡沫浸渍法[10]。将NaOH 溶液改性后的PU 海绵完全浸入由20 mL 的无水乙醇溶剂、56wt%的基础玻璃粉末,1.8wt%的乙基纤维素(ethyl cellulose ethoce,EC),5 mL 15wt%的聚乙二醇(polyethylene glycol,PEG)配制成的浆料中,用手将其压于烧杯内壁排出过多浆料。然后在室温下干燥24 h 后置于鼓风干燥箱中(设定程序80℃,2 h),将其放入高温箱式电阻炉中在Ts 与Tc 范围内试烧,确定烧成温度,最终温度见表5(300℃为PU海绵完全挥发的温度),随炉冷却,制得试件(示意图见图3)。测试之前,去除有堵孔、坍塌、破损等有缺陷的试件。F组玻璃支架在烧成过程中全部塌陷,故后期性能测试排除F组分的玻璃支架。
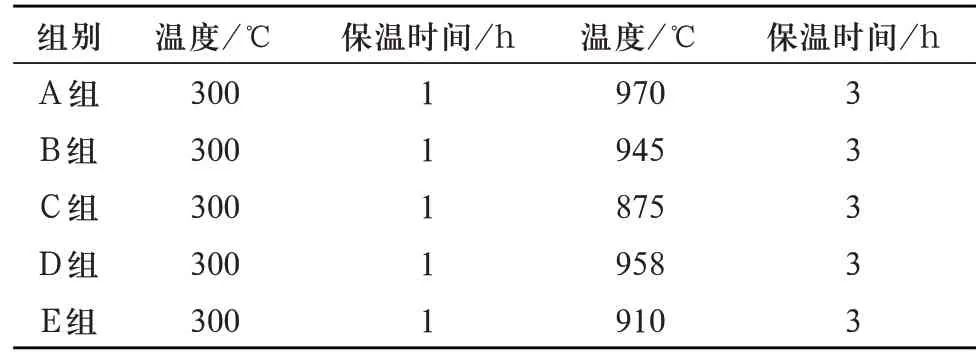
表5 多孔生物活性玻璃支架的烧成温度

图3 多孔支架三种规格试件示意图
1.3.4 标准模拟体液的配制 为模拟植入环境,测试试件的生物活性,配制标准模拟体液(simulated body fluid,SBF)(表6、表7)。
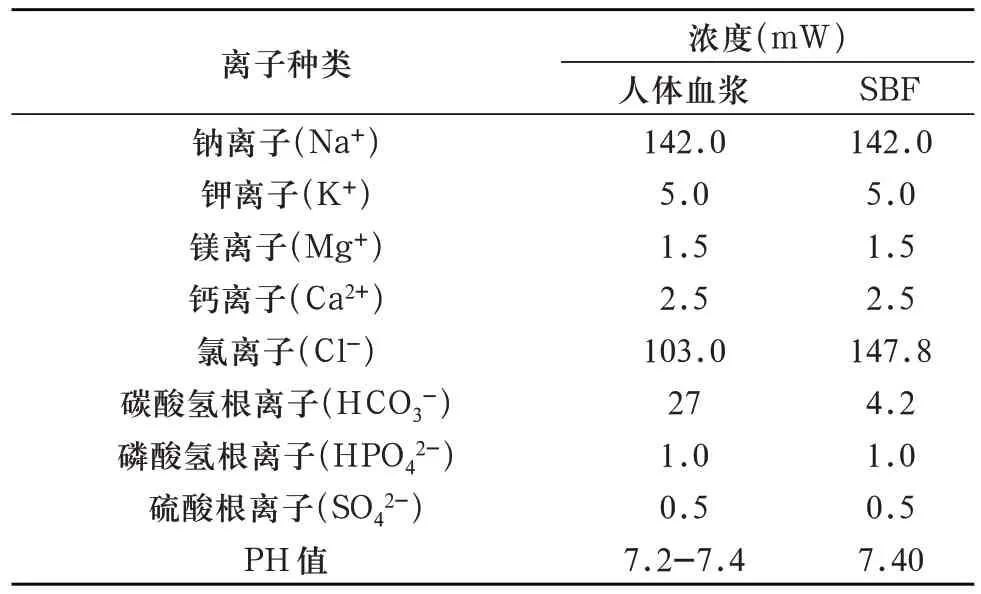
表6 SBF与人血浆各离子的浓度异同
1000 mL SBF 溶液按以下步骤配制[11,12]:聚乙烯烧杯(内壁光滑无痕,因磷灰石会在玻璃器皿和划痕处成核)盛有700 mL 去离子水放入带有恒温水浴功能的磁力搅拌机中,定温至36.5℃±0.5℃。将表7 的试剂按①-⑧依序称量溶解到去离子水中(不可一同溶解几种试剂,完全溶解后再加下一种。试剂③④⑤⑦⑧暴露于空气中易吸潮,称量时使用称量瓶密封)。
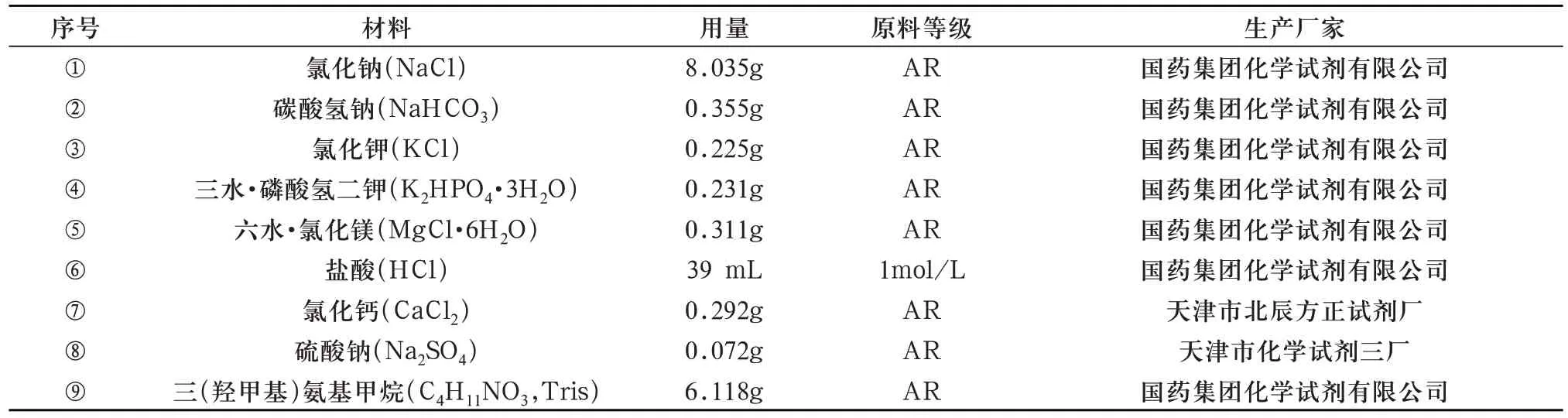
表7 配制SBF溶液所需试剂
最后一个试剂按以下方法溶解:
待①-⑧号试剂完全溶解后,加去离子水至900 mL,恒定溶液温度至36.5℃±0.5℃,pH计测试,此时pH值应在2.0±1.0范围内。将Tris分成几小份逐份加入,同时关注pH值变化,每次待上一份完全溶解并pH值稳定后再加下一份(一次加入Tris的量不可过大,否则pH值剧烈变化易引起磷酸钙沉淀)。当pH值接近7.45时,滴加1 M盐酸将pH值调至7.42,继续溶解,过程中pH值保持在7.42-7.45间,温度恒定36.5℃±0.2℃,最后调节pH值至7.40。
移除清洗pH 计电极及搅拌子,洗液收入溶液中,温度降至20℃后定容至1000 mL,密封于塑料瓶中,冰箱4℃冷藏,保质期1个月。注意配制成功的SBF要无色透明且烧杯内不能有沉积,配制过程中,一旦出现沉淀则表示配制失败。
1.3.5 多孔生物活性玻璃支架抗压和抗弯强度的测试 利用万能力学试验机在室温干燥环境下进行测试,结果取每组5个试件的平均值。单轴压缩试验压10 mm×10 mm×10 mm的立方体试件测定其抗压强度(compressive strength,CS)。三点弯曲试验压25 mm×5 mm×5 mm的长条形试件测定其抗弯强度(bending strength,BS)。参照JIS R1601 标准[13],加载速率0.5 mm/min,最大载荷5.0kN。试样的抗压强度按照公式1 进行计算。

式中: σ——抗压强度,MPa
P——试样的破坏载荷,N
S——样品与探头的接触面积,mm2
试样的抗弯强度按照公式2进行计算。

式中:σ——抗弯强度,MPa
F——最大弯曲载荷,Nl——跨距,mm
b——试样的宽,mm d——试样的高,mm
1.3.6 多孔生物活性玻璃支架降解性能的测试 多孔生物活性玻璃支架的降解性能通过体外浸泡实验进行测试,首先,将试件浸泡在无水乙醇中于超声波清洗机中荡洗5 min,自然风干。称浸泡前干重,以试样质量与溶液体积比为1.0 mg/mL的比例将每组各5个5 mm×5 mm×5 mm 试件浸泡在装有SBF 的聚乙烯瓶中。置于37℃的恒温水浴箱中,每24 h 更换一次溶液。分别在1 d、3 d、7 d 三个时间段取出试件,去离子水和无水乙醇洗净后用鼓风干燥箱80℃烘干,称重。按公式3 计算质量损失百分比。
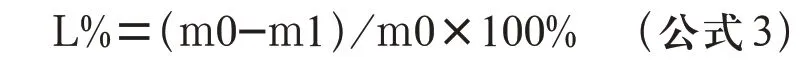
式中:L%——质量损失百分比
m0——浸泡前样品干重,g
m1——浸泡后样品干重,g
1.3.7 多孔生物活性玻璃支架体外矿化活性的测试 浸泡实验同降解测试,但区别是无需更换SBF 浸泡液,在相同时间取出、冲洗、干燥试件。选取支架表面平整部分制成片状试件,贴导电胶、喷金以增强试件导电性。随后利用扫描电镜选取5个随机位置拍照记录各组试件浸泡后的形貌与微观结构的变化;用三头研磨机将未喷金试件磨细成粉后,X 线衍射分析支架浸泡前后物相组成[14,15],对于是否有体外羟基磷灰石生成的确认基于粉末衍射标准联合委员会(JCPDS-No.09-0432)[16]。
1.4 统计学方法 使用SPSS 24.0 软件对所得数据进行统计学分析,计量资料用(±s)表示,若数据满足方差分析条件(正态分布,总体方差齐),采用单因素ANOVA 检验比较多组数据结果,事后多重比较用LSD 检验,若数据不满足方差分析条件,采用秩和检验,P≤0.05 为差异有统计学意义。
2.结果
2.1 P2O5含量对多孔生物活性玻璃支架抗压强度的影响 对A、B、C、D、E 五组多孔生物活性玻璃支架单轴压缩测试所得数据进行单因素ANOVA 检验,可得五组支架抗压强度有统计学差异(F=367.912,P=0.00<0.05)。进一步LSD多重比较,除A组与B组之间无统计学差异,其余各对比组之间均有统计学差异(见表8)。说明随着支架P2O5含量超过1wt%并逐渐增加,抗压强度逐渐增强。
表8 A、B、C、D、E五组试件抗压强度(MPa)对比(±s)
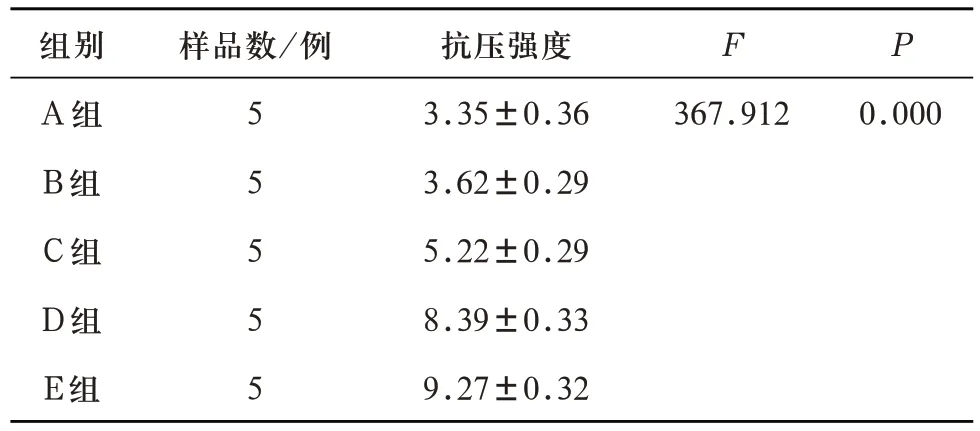
表8 A、B、C、D、E五组试件抗压强度(MPa)对比(±s)
注:A-B组两两比较P=0.197,A-C组、A-D组、A-E组、B-C组、B-D组、B-E组、C-D组、C-E组、D-E组两两比较均P=0.000
2.2 P2O5含量对多孔生物活性玻璃支架抗弯强度的影响 对A、B、C、D、E 五组多孔生物活性玻璃支架三点弯曲测试所得数据进行单因素ANOVA 检验,可得五组支架抗弯强度有统计学差异(F=244.041,P=0.00<0.05)。进一步LSD 多重比较,除A组与B组之间无统计学差异,其余各对比组之间均有统计学差异(见表9)。说明随着支架P2O5含量超过1wt%并逐渐增加,抗弯强度逐渐增强。
表9 A、B、C、D、E五组试件抗弯强度(MPa)对比(±s)

表9 A、B、C、D、E五组试件抗弯强度(MPa)对比(±s)
注:A-B组两两比较P=0.677,A-C组、A-D组、A-E组、B-C组、B-D组、B-E组、C-D组、C-E组、D-E组两两比较均P=0.000
2.3 P2O5含量对多孔生物活性玻璃支架降解性能的影响 对A、B、C、D、E 五组多孔生物活性玻璃支架1 d、3 d、7 d 降解性能测试所得质量损失百分比分别进行分析。浸泡1 d 后,五组多孔支架的质量损失百分比有统计学差异(F=35.177,P=0.00<0.05)。进一步LSD 多重比较,除A、B、C 对比组相互之间质量损失百分比无统计学差异,C组和D组之间质量损失百分比无统计学差异,其余各对比组之间的质量损失百分比均有统计学差异(见表10)。说明在浸泡实验初期,高磷含量组(D组、E组)降解性能强于低磷含量组。
表10 A、B、C、D、E五组试件浸泡实验1 d的质量损失百分比(%)对比(±s)
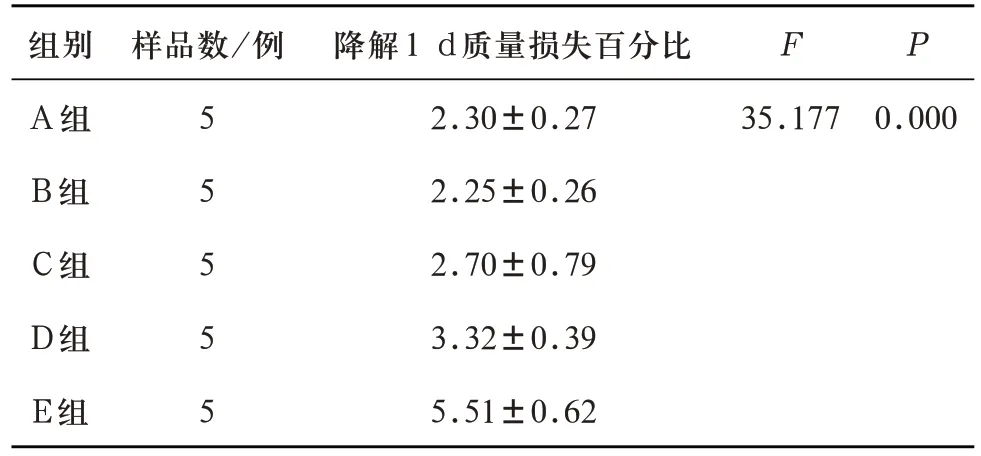
表10 A、B、C、D、E五组试件浸泡实验1 d的质量损失百分比(%)对比(±s)
注:A-B组两两比较P=0.888,A-C组两两比较P=0.227,A-D组两两比较P=0.005,B-C组两两比较P=0.180,B-D组两两比较P=0.004,C-D组两两比较P=0.070,A-E组、B-E组、C-E组、D-E组两两比较均P=0.000
浸泡3 d 及7 d 后,五组多孔支架的质量损失百分比差异有统计学意义。进一步LSD多重比较,除A组、B组之间的质量损失百分比无统计学差异,其余各对比组之间的质量损失百分比均有统计学差异(见表11、表12)。说明随着多孔生物活性玻璃支架内P2O5含量超过1wt%并逐渐增加,降解性能逐渐增强。
表11 A、B、C、D、E五组试件浸泡实验3d的质量损失百分比(%)对比(±s)
表11 A、B、C、D、E五组试件浸泡实验3d的质量损失百分比(%)对比(±s)
注:A-B组两两比较P=0.955,A-C组两两比较P=0.007,B-C组两两比较P=0.008,A-D组、A-E组、B-D组、B-E组、C-D组、C-E组、D-E组两两比较均P=0.000
表12 A、B、C、D、E五组试件浸泡实验7 d的质量损失百分比(%)对比(±s)

表12 A、B、C、D、E五组试件浸泡实验7 d的质量损失百分比(%)对比(±s)
注:A-B组两两比较P=0.964,A-C组、A-D组、A-E组、B-C组、B-D组、B-E组、C-D组、C-E组、D-E组两两比较均P=0.000
以上结果说明,当生物玻璃中磷含量为1wt%时与不掺杂磷的玻璃并无明显的降解性能差异,当磷掺杂量达到3wt%并逐渐增多,玻璃的降解性能逐渐增强(见图4)。
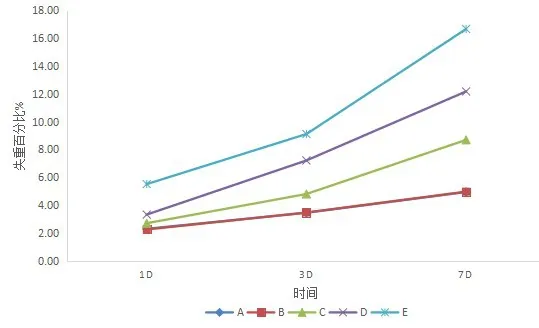
图4 A、B、C、D、E五组试件浸泡实验不同时间的失重曲线
2.4 P2O5含量对多孔生物活性玻璃支架矿化性能的影响 图5为经体外浸泡实验的A、B、C、D、E五组试件的扫描电镜(SEM)照片。比较几张图片可见,在标准模拟体液中浸泡7 d后,A组、B组试件表面只有少量甚至没有颗粒状物质沉积。C组在浸泡7 d后试件表面可见明显颗粒状物质出现。D组试件在浸泡1 d后表面即可有明显的颗粒沉积,浸泡3 d后表面可见大量的颗粒状物质,浸泡7 d后表面物质相互融合形成薄膜样沉积。E组试件在浸泡1 d后表面就沉积大量的颗粒状物质,3 d后表面的颗粒状物质相互融合形成薄膜样沉积,7 d后表面的薄膜即相互融合成片,基本覆盖整个试件表面。

图5 A、B、C、D、E五组试件浸泡实验后的SEM照片
对标准模拟体液浸泡实验1 d、3 d、7 d 后样品的X 线衍射分析(XRD)也为扫描电镜结果提供了有力的佐证(图6)。A组、B组试样浸泡7 d后的图谱与浸泡前相似,仍为馒头峰,说明试件表面沉积的颗粒并不是结晶态的羟基磷灰石,物相组成无明显差异。C组浸泡前与浸泡3 d后图谱均为馒头峰,浸泡7 d 后,图谱上出现了代表羟基磷灰石的(002)晶面和(211)晶面的特征衍射峰,说明表面的沉积物为结晶态的羟基磷灰石。浸泡前D 和E组试件的图谱为馒头峰,浸泡1 d 后图谱上即出现了代表羟基磷灰石特征衍射峰。说明随支架中P2O5含量增加,其体外矿化性能逐渐增强。
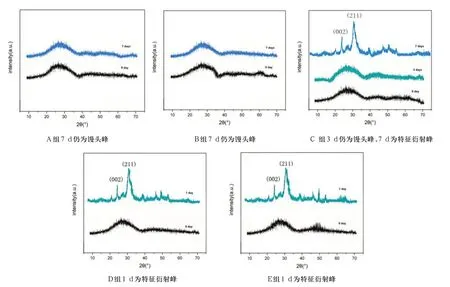
图6 A、B、C、D、E五组试件浸泡实验后的XRD图谱
3.讨论
3.1 P2O5对生物活性玻璃力学性能的影响机制 在本实验中生物活性玻璃主要由SiO2作为网络形成体构成结构的最基本单位,在玻璃网络中表现为SiO4四面体。多个四面体顶角相对,形成三维支架样结构。四面体中的O 原子被称为桥氧,通过名为桥氧键的离子键连接2个Si原子。当玻璃组分中添加了网络修饰体Na2O,因为它是低电负性阳离子的金属氧化物,无法建立玻璃网络结构,所以会通过破坏桥氧键,形成1个O 原子连接1个Si 和另1个非硅原子的非桥氧键来掺入网络结构(如图7所示)。相比于桥氧键,非桥氧键要弱得多,无法维持原有的SiO4四面体形成的三维结构,导致空间网络发生解聚,三维结构变为二维,最终使玻璃的力学性能降低[17,18]。掺入P2O5恰好逆转了这种趋势,因为P2O5同是作为网络形成体参与构建玻璃网络,且Na 对磷酸根的亲和力更高,磷酸根离子可以把Na 改性剂从硅酸盐网络中剥离出来形成正磷酸钠,使被Na2O 破坏掉的Si-O-Si 桥氧键重新连接起来,优化网络连接性,使玻璃力学性能提高。故在本实验中,当P2O5含量达到3wt%并逐渐增加,支架的抗弯强度和抗压强度逐渐增强。但当基础玻璃中P2O5的含量达到12wt%时在试件烧制过程中支架全部塌陷,无法烧制成型。原因是随着P2O5含量增多,有一部分的P-O-Si 键逐渐生成,玻璃的三维结构网络变为二维,会使玻璃的力学性能有降低的趋势,当这种趋势累积到一定程度,再加入P2O5会使玻璃的力学性能转而下降[7]。此外,退火过程也会影响生物活性玻璃支架的力学性能。本实验制备玻璃支架时需要将依附在海绵骨架上的玻璃粉末再加热至软化温度与析晶起始温度之间,经历了退火过程后,玻璃的力学性能发生了改变,但不同于晶体材料退火后力学性能降低,生物活性玻璃的力学性能反而提高,因为对于晶体材料而言,经历退火后晶体密度会因键密度(结构密度)的增加而增加,降低材料的力学性能。而玻璃的退火增加了结构密度而不影响玻璃密度,因此,退火也可导致玻璃的力学性能增强[19]。
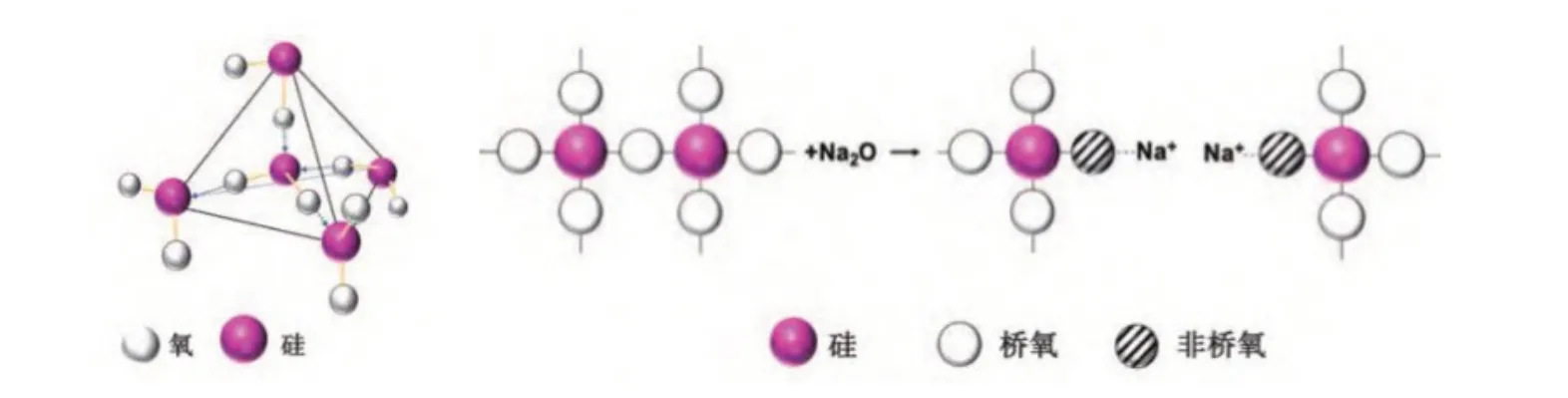
图7 玻璃结构示意图
3.2 P2O5对生物活性玻璃生物活性的影响机制在本实验中,当生物玻璃中磷含量为1wt%时与不掺杂磷的玻璃并无明显的降解性能差异,体外矿化活性较低。当磷掺杂量达到3wt%并逐步增多,玻璃的生物活性随之增强。有研究表明,采用高温熔融工艺不可制备SiO2含量超过60%的生物活性玻璃,否则无生物活性,植入人体后不会与骨骼产生结合[20]。具有理想生物活性的生物活性玻璃植入人体后,体液中的氢离子与材料发生反应,材料自身的钠离子与钙离子被迅速置换出来,引起材料的硅氧键断裂重新合成Si-OH,并在材料表面发生聚合反应形成一层凝胶层,此时因体液环境中的氢离子发生置换反应数量减少,植入处呈弱碱性使材料表面凝胶层电荷表现为负,可吸附Ca2+和PO43-形成CaO-P2O5层,最终结合体液中CO32-和OH-生成HCA[21]。但在此过程中,新形成的CaO-P2O5层是不稳定的,很快就被溶解了,仅由持久的硅酸盐网络组成的颗粒核心保留下来。导致羟基磷灰石不易生成。而对于基质中含磷的基础玻璃,一旦形成了CaO-P2O5层,该组颗粒会迅速生长形成稳定的羟基磷灰石[22]。
在早期,玻璃网络聚合度因与玻璃溶解度之间有着的密切关系,故一直将其视为生物活性玻璃体外生物活性的潜在重要因素,认为玻璃高的降解性能可通过快速增加局部微环境内羟基磷灰石的生成所需的原料来提高矿化性能[7]。但后期研究发现,生物活性玻璃中掺杂的磷不仅能直接通过此种途径来提高矿化活性,还能通过降低其对精确的硅酸盐网络连通性的依赖性来促进磷灰石的形成,从而实现高且恒定的体外生物活性,即当玻璃内含足够高的P2O5含量(≥4mol%)时,随着网络聚合度的增加,玻璃溶解度降低不会对玻璃的生物活性产生不利影响。经探究尽管随着磷酸盐含量的增加生物活性玻璃的硅酸盐网络变得越来越紧密,不易水解,但与此同时,玻璃中游离正磷酸盐基团的总量也会增加,其释放对生物活性产生积极影响,由于游离磷酸盐可用性更高,其产生的积极影响最初克服了硅酸盐网络变紧密的不利影响。因此,磷的掺杂对玻璃产生两个相反的结构作用,其平衡将决定该成分玻璃的生物活性最终会增强或减弱[7,23]。
此外,生物活性同样与玻璃支架和SBF溶液接触面积有关。但一项研究发现随着玻璃中P2O5含量的增加,玻璃支架与SBF溶液的接触面积对于体外生物活性的影响逐渐减弱:P2O5含量最低(2.6 mol%)的“小颗粒”粉末(直径<50 μm)样品在SBF中浸泡24 h后没有形成羟基磷灰石,而“大颗粒”粉末(直径50-100 μm)则显著生成羟基磷灰石,形成鲜明对比。但P2O5含量高(6.0 mol%)的玻璃中两个粒子直径产生的羟基磷灰石的量却非常相似,这些结果有力地表明,磷含量高的玻璃其体外生物活性对于玻璃与SBF溶液接触的表面积的依赖降低[23]。
4.结论
在一定掺入量范围内,P2O5替代SiO2会加强多孔生物活性玻璃支架的抗压强度和抗弯强度,本实验中当P2O5含量达到12wt%时支架无法烧制成型,全部塌陷。P2O5替代SiO2可加强多孔生物活性玻璃支架的降解性能及体外矿化活性。为后续进行成骨细胞体外培养及活体内生物相容性实验提供前期参考,为将来新型生物活性玻璃骨组织修复材料的研制提供一定的理论支持。
