短链脂肪酸调节肠黏膜屏障缓解炎性肠病新进展
2022-11-26郭雪然贺程威华嵘暄杜奕萱尚宏伟徐敬东
郭雪然,贺程威,高 晗,华嵘暄,梁 宸,杜奕萱,尚宏伟,路 欣,徐敬东
郭雪然,梁宸,首都医科大学2019级临床医学专业 北京市 100069
贺程威,高晗,徐敬东,首都医科大学基础医学院生理学与病理生理学系 北京市 100069
华嵘暄,首都医科大学2020级临床医学“5+3一体化” 北京市 100069
杜奕萱,首都医科大学2020级口腔医学专业“5+3一体化” 北京市100069
尚宏伟,路欣,首都医科大学基础医学院形态学教学实验室 北京市100069
0 引言
随着人类对肠道黏膜免疫系统以及炎症性肠病(inflammatory bowel disease,IBD)易感性的研究的不断完善,越来越多的证据表明肠道微生物在调节人体健康中发挥着关键作用.宿主和肠道菌群相互作用,共同维持肠黏膜屏障的功能,调节肠道内环境稳态[1],当肠道微生物中致病菌和正常菌群比例失调[2],致病菌增多可导致肠上皮通透性增高、黏膜免疫失调、影响肠上皮细胞(intestinal epithelial cells,IECs)的能量代谢与正常IECs结构和功能,诱发肠道炎症.目前,肠道微生物代谢产物与宿主之间的相互调节作用成为科学家们研究的焦点.肠道微生物代谢产物,特别是短链脂肪酸(short-chain fatty acids,SCFAs)能够促进肠黏膜屏障对多种微生物信号的整合.肠黏膜屏障通透性改变、功能障碍以及微生物群失调等均是IBD等炎症性肠道疾病的诱因,而探索其作用机制目前成为了研究热点.本篇综述将就SCFAs通过调节肠黏膜屏障来缓解IBD的作用机制做出阐述.
1 IBD患者的肠道菌群及其代谢产物SCFAs
IBD是累及回肠、直肠、结肠的一种特发性肠道炎症疾病,以慢性炎症反应和胃肠道上皮为主要特征,主要包括溃疡性结肠炎(ulcerative colitis,UC)和克罗恩病(Crohn’s disease,CD)[3].众所周知,IBD的发生可能与遗传、免疫、微生物和环境等多重因素相互作用有关[4],其发生机制尚不明确,但普遍认为,IBD的发生可能与个体肠道内菌群失调有关.肠道菌群能够抵抗病原菌的侵袭,当肠道菌群和黏液层之间的稳态受到外来因素的影响时,引起肠黏膜屏障功能的改变以及黏膜伤口的出现.肠上皮屏障功能受损及黏膜伤口愈合不足或延迟是IBD发展和持续的因素.肠黏膜屏障能使营养物质选择性通过并抵抗有害物质进入,黏膜伤口的修复由上皮细胞和浸润型免疫细胞及其介质的动态串扰进行协调,参与修复、增殖和分化等复杂机制的调控,还依赖于免疫应答、介质之间的相互作用等一系列事件进行调控[5,6].通过重建肠黏膜屏障、调控其转运作用并促进黏膜伤口的愈合来治疗IBD或将成为亟待开发的新疗法[7].
新一代测序技术的蓬勃发展使人们更易识别IBD患者中肠道微生物群组成的变化.利用宏基因组测序可以揭示微生物群的功能潜力.IBD患者的粪便微生物中,拟杆菌属、厚壁菌属、普拉氏梭菌和人罗斯拜瑞氏菌减少,而放线菌属和变形杆菌属增加,促炎细胞因子水平升高,促进肠道炎症的发展[8,9];在黏膜组织和粪便采样中,CD患者的巴斯德杆菌、韦荣氏球菌、奈瑟氏菌、梭杆菌属和大肠杆菌科丰度增加,拟杆菌属、梭菌目、粪杆菌属、罗氏菌属、布劳特氏菌属、瘤胃球菌和毛螺菌科的丰度降低,CD患者发酵途径减少,糖降解率升高,醌类生物合成增加形成了具有炎症特征的微生物环境[10].
利用元转录组学发现,在基因组学上丰富的物种更倾向于贡献更多的转录途径,揭示了肠道菌群的功能活性.IBD患者的鲁米诺球菌丰度在RNA水平显著增加,而在DNA水平仅轻度增加[11];哈氏梭菌、鲍氏梭菌丰度明显增加,提示它们的作用比单一基因组学丰度的差异更明显[12].
肠道菌群代谢产物对维持细胞正常的生理活动具有重要意义.其中,SCFAs又称挥发性脂肪酸,是由1-6个碳原子组成的有机脂肪酸,主要由肠道中残留的难以消化的膳食纤维在肠腔中经厌氧菌发酵产生,大多由膳食纤维在结肠腔内经细菌酵解生成,也有少量来自于膳食蛋白和内源性蛋白.短链脂肪酸种类多、功能复杂,引起广泛关注.乙酸、丙酸和丁酸是肠内主要的短链脂肪酸,约占所有SCFAs的95%以上,其在肠道中的摩尔比为60:60:20,在肠腔中达到峰值浓度(50 mM-100 mM)[13-15].据估计,近端结肠总量约70 mM-140 mM,远端结肠总量约20 mM-70 mM[16-18].如表1所示,它是 IECs重要的能量来源[19],可通过多种机制抵抗病原体感染和应激损伤,包括诱导抗菌肽LL37的表达、调节性T细胞(regulatory T cell,Tregs)的增殖分化、激活G蛋白偶联受体41(G protein-coupled receptor 41,GPR41)、GPR43和GPR109a以及激活NOD样受体蛋白3(NOD-like receptor thermal protein domain associated protein 3,NLRP3)来抵御病原体感染及应激损伤[20].代谢组学研究表明在IBD患者中,肠道菌群紊乱导致短链脂肪酸减少.IBD患者的丁酸和丙酸水平显著下降,给予丁酸酯和丙酸酯可有效缓解IBD的相关症状,提示丁酸和丙酸在IBD的发生中具有一定的作用[21].临床研究表明[22],使用丁酸灌肠后患者症状明显改善,同时发现白细胞计数、红细胞沉降率、核因子κB(nuclear factor kappa-B,NF-κB)以及白细胞介素-1β(interleukin-1β,IL-1β)的水平均显著下降[23],这些结果进一步表明SCFAs产生菌或SCFAs自身通过影响肠道免疫反应、基因表达、细胞增殖以及宿主代谢等作用于宿主细胞,引起肠道炎症[16,24,25].尤其是产丁酸菌的减少引起丁酸浓度降低,导致异常免疫应答,引起黏膜损伤,从而使黏膜下层发生非特异性炎症反应.

表1 与SCFAs相关的肠道菌群在IBD中的变化与功能
IBD包括UC和CD两种疾病,由于发病机制与表现的不同,SCFAs在其中具有不同的作用.就保护肠黏膜屏障方面,UC与CD结肠中粘液层均变薄,MUC2产生减少,丁酸可通过刺激MUC2[26]表达从而产生黏蛋白保护肠粘液屏障以及增强ITF[18]改善肠黏膜伤口愈合.而CD病人的发病因素还包括Paneth细胞功能的障碍以及肠道细胞抗菌能力减弱[27],NOD2是CD病人最显著的遗传风险因素之一,它由Paneth细胞、树突状细胞、巨噬细胞和吸收性IEC表达,当α-防御素水平较低时,NOD2可能发生突变,导致抗菌功能受损[28].CD病人中,SCFAs还能增加紧密连接蛋白表达降低肠上皮通透性[29],通过GPCR激活NLRP6[30],促进肠杯状细胞分泌粘液,促进肠道免疫,逆转结肠炎.此外,SCFAs对UC和CD病人均具有促进肠黏膜通透性与维持水电解质交换平衡的作用.就影响宿主免疫应答反应方面,二者作用途径类似.SCFAs作用于多种免疫细胞调控其分化、趋化、募集、增殖和凋亡等过程,同时减少白细胞介素1(interleukin-1,IL-1)、白细胞介素6(interleukin-6,IL-6)、肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)等促炎因子的产生,抑制NF-κB的转录与迁移;通过叉头转录因子(forkhead winged transcription factor,Foxp3)乙酰化水平增加,作用于GPR43来诱导Treg细胞增殖分化,促进粘蛋白(mucins,MUC)、抗菌肽(antimicrobial peptides,AMP)以及细胞因子的分泌,通过GPR41和GPR43激活NLRP3,促进IL-1β和白细胞介素18(interleukin-18,IL-18)的分泌[31,32];通过HDAC抑制途径使组蛋白乙酰化,抑制NF-κB途径;抑制γ-干扰素(interferon-γ,IFN-γ)/STAT1信号通路,诱导T细胞凋亡[33].
目前,多项临床研究证明,补充丁酸可以有效缓解IBD,主要应用于辅助治疗,而丁酸等SCFAs的作用机制还有待进一步明确.
2 短链脂肪酸在肠上皮细胞的吸收、代谢与供能
肠黏膜屏障是机体吸收养分的主力军,能整合多种微生物信号抵御病原体的入侵,协调肠腔微生物和宿主免疫系统之间的相互作用,维持宿主-微生物和组织内稳态,促进肠道菌群和宿主共生[1].肠黏膜屏障分为由上皮细胞层和黏膜层组成的物理屏障、由免疫细胞和肠黏膜分泌的抗体组成的免疫屏障和由肠道共生菌组成的生物屏障[43].任意一种屏障功能的缺陷均可能导致有害因子入侵,机体免疫系统过度活化,引发黏膜炎症,而导致IBD.
SCFA在肠道中的吸收经历了一个复杂的过程,它多以SCFA-的形式存在,通过多种方式被肠道上皮细胞摄取(如表2),包括被动扩散、通过离子通道转运(如图1A)以及通过G蛋白偶联受体(G protein-coupled receptor,GPCR)介导(如图1B和C)进行运输[44].
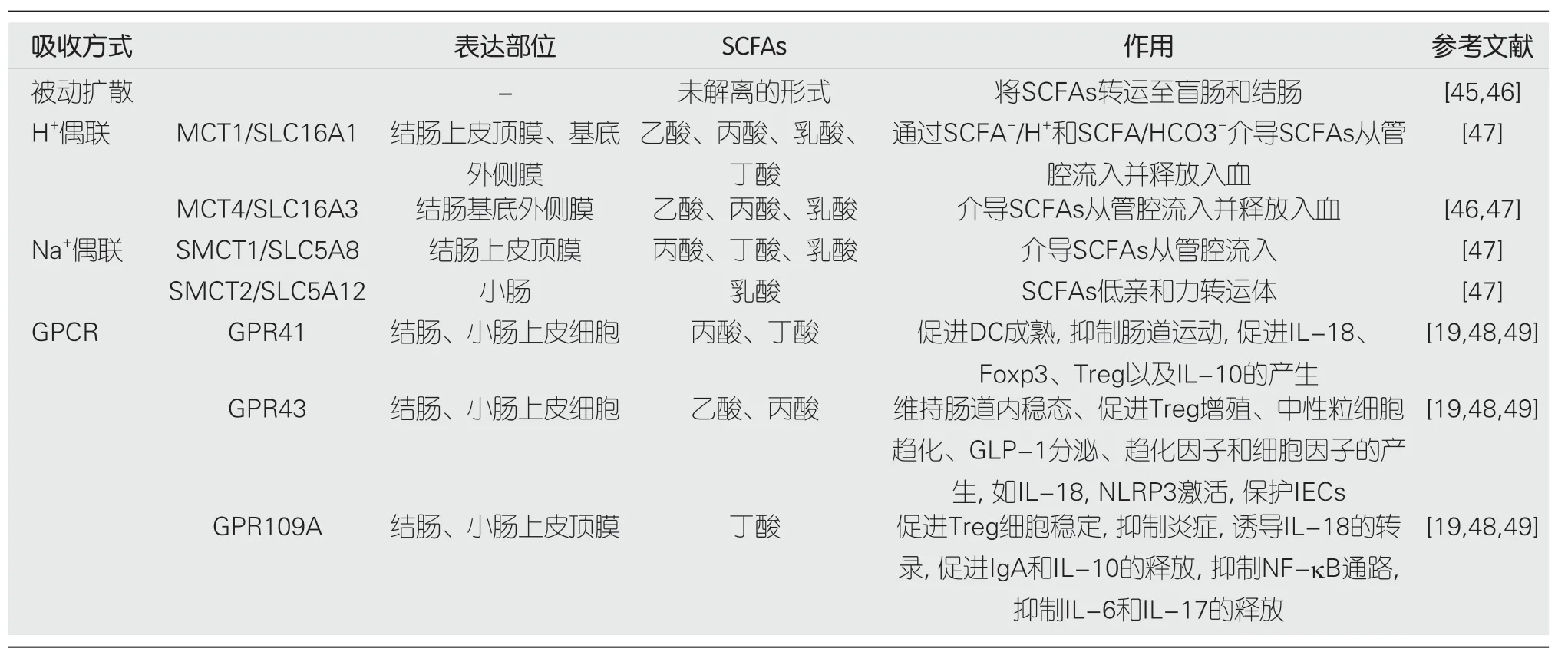
表2 SCFAs在肠道上皮细胞的转运
载体介导的途径在SCFAs摄取的过程中发挥了重要的作用(如图1A).SMCT1(SLC5A8)和SMCT2(SLC5A12)是Na+偶联的载体,SCFAs--HCO3-相偶联通过SMCT1介导带电的转运(Na+:SCFAs-=2:1),SMCT2介导电中性的转运(Na+:SCFAs-=1:1),MCT1(SLC16A1)和MCT4(SLC16A3)是H+偶联的载体,介导SCFA的电中性转运(H+:SCFAs-=1:1)[47].SCFAs在细胞内外都能调节黏膜免疫反应、维持肠黏膜屏障功能以及刺激黏膜生成.在细胞外,SCFAs主要通过结肠微绒毛膜上的受体发挥作用,在细胞内则通过抑制组蛋白去乙酰化酶(histone deacetylases,HDAC)、产生能量并转化为酮体参与细胞能量代谢[47].
肠道炎症及促炎因子对MCT1的表达和功能具有调控作用[50].使用药物诱导小鼠产生肠炎,发现小鼠结肠上皮中MCT1基因及其蛋白表达量显著下降,在IECs培养基中加入IFN-γ和TNF-α后,通过QT-PCR、免疫印迹和免疫组化证明MCT1的mRNA和蛋白质水平显著降低,丁酸的转运能力降低[51],MCT1表达量下调与WNT/β-catenin通路活性降低有关[52],该通路通过氧化应激调控炎症.提示肠道炎症中丁酸氧化不足是MCT1介导的丁酸摄取减少的结果.
特别值得关注的是SLC5A8,有研究表明丁酸盐通过SLC5A8将肠道菌群、膳食纤维和黏膜免疫系统联系起来[53,54].给野生型(wild type,WT)小鼠和Slc5a8-/-小鼠分别喂食最佳纤维摄入饮食(fiber-containing diet,FC-diet)和无纤维摄入饮食(fiber-free diet,FF diet),对于摄入FC饮食的小鼠的存活率、直肠出血和腹泻等症状以及结肠炎的进展均无显著差异.而对于摄入FF饮食的小鼠,普雷沃菌、萨特氏菌、丹毒丝菌的丰度较FC饮食的小鼠高,Slc5a8-/-小鼠的小肠和结肠出血严重、体温低,其结肠固有层有轻度炎症和免疫细胞浸润,小鼠体内粘液分解的艾克曼菌含量更高,这可能导致结肠粘液层变薄,患结肠炎风险增高.此外,Slc5a8-/-小鼠在经DSS治疗后表现出急性发病.通过结肠上皮转录组的分析,在Slc5a8-/-小鼠中,有助于黏膜修复的缺氧诱导因子(hypoxia-inducible factor-1α,HIF-1α)表达降低,而SCFAs作为能量底物,通过SLC5A8与HIF-1α结合,并增加其稳定性[53,55].此外,Slc5a8-/-小鼠的预防结肠炎的Akt信号途径活性降低,Tlr2表达受抑制,从而使黏液三叶因子(trefoil factor,ITF)中的Tff1和Tff3的表达下调[56],调节性免疫细胞被抑制从而诱发炎症[57].综上所述,SCFAs的转运在IBD的发生和治疗中发挥着重要的作用,对SCFAs的转运通道的调控与肠黏膜的保护作用、肠上皮细胞的能量代谢作用以及维持肠黏膜稳态等均密切相关.
3 SCFAs保护肠上皮细胞
众所周知结肠上皮由多种细胞构成,形成了一个精细的防护屏障.SCFAs进入肠上皮细胞后,能够通过一系列的信号通路进入不同的细胞结构中,其中最受关注的是线粒体和细胞核,通过调控基因的转录与表达以及炎性因子的释放,有助于促进肠上皮细胞的增殖,并保障其完整性.
3.1 SCFAs促进肠上皮细胞增殖 结肠上皮细胞群是一个动态的群体,从干细胞到最终分化的结肠细胞,保持着增殖、分化、凋亡、免疫反应之间的动态平衡,其中,有一些关键的调节因子在精准调控这一过程,其中肠道菌群及其代谢产物SCFAs是这一过程中不可或缺的重要参与者.
研究发现[58],当结肠内SCFAs减少,上皮细胞的增殖受抑制,给大鼠结肠分别灌注乙酸、丙酸、丁酸后,结肠上皮细胞的增殖明显的改善,其中丁酸作用效果最强.通过体外培养[59,60],在Wistar大鼠的移植小肠上补充丁酸,10 d之后在透射电镜下观察发现,经丁酸处理肠黏膜绒毛高度、隐窝深度和黏膜厚度均显著高于对照组,提示丁酸对肠黏膜有明显的促生长作用.
SCFAs维持肠黏膜稳定性的作用可能与AP-1信号通路激活机制有关[61].丙酸盐和丁酸盐均是AP-1途径的有效激活剂,尤其丁酸盐的激活效率更高,它们可以增强PMA诱导的c-fos和ERK1/2磷酸化表达,减弱细胞周期蛋白D1[62],降低与细胞增殖相关的其他基因水平,如cdk1以及诱导分化的特异性基因,包括转谷氨酰胺酶Ⅰ型.其中,ERK1/2主要与细胞生长、增殖和分化有关,在HT-29和Caco-2细胞中[61],发现丁酸盐单独作用对ERK1/2磷酸化有轻微的影响,但是丁酸盐与PKC激活剂PMA一同作用显著地促进ERK1/2的磷酸化,提示丁酸盐与PMA共同使用对AP-1途径具有协同的作用.ERK1/2的过度诱导有助于激活c-fos,该基因也被认为是ERK1/2激活持续时间的“传感器”[63].因此,SCFAs不仅能够作为肠上皮细胞的主要能量来源,还能够促进上皮细胞增殖、分化,减少细胞凋亡,对维持肠黏膜上皮细胞的稳定性具有重要意义[64].
3.2 SCFAs保护肠黏膜屏障的完整性 肠道黏膜屏障在宏观和微观上的完整性在维持肠道稳态中具有重要的意义,SCFAs可以通过调节炎性因子的释放以及肠上皮细胞的自噬等来维持肠黏膜的完整性.
SCFAs与肠上皮细胞GPR43受体结合,刺激K+的流动,使肠上皮细胞膜超极化,通过调节活性氧的生成激活NLRP3炎性小体[65],促进IL-1的分泌,并且通过激活Caspase 1,增加pro-IL-18的表达[66],促进IL-18的产生,保持肠上皮细胞的完整性[67].在DSS诱导的结肠炎小鼠的结肠上皮细胞显示[67],在高膳食纤维喂养或经醋酸盐处理后的WT小鼠血清中IL-18水平远高于Gpr43-/-和Gpr109a-/-小鼠,同时Gpr43-/-小鼠和Gpr109a-/-小鼠未能表现出体重增加、结肠长度的改善或是结肠组织损伤的减少,说明GPR43和GPR109a参与到了IL-18分泌的过程中,并改善IBD.体内IL-18的释放与一些肠道菌群相关,通过Pearson相关性,丁酸产生菌Lachnospiraceae[68]、乙酸和丙酸产生菌Rikinellaceae和Porphyromonadaceae[69]与SCFAs相关,且与Myd88[70]、Irak[71]、TLR5[72]和TLR4[73]有关,提示可能参与TLR信号通路.
除此之外,特别值得关注的是GPR43是SCFAs在中性粒细胞上的唯一受体,参与炎性因子的释放以及调节O-的产生和吞噬作用.中性粒细胞聚集可使肠黏膜屏障受损,加重IBD的症状.在大鼠体外模型中[74],发现SCFAs可增加中性粒细胞地迁徙和黏附,还能提高中性粒细胞表面L-selectin mRNA的水平与表达,SCFAs是通过GPR43参与中性粒细胞的趋化反应中与稳态调节,实现机体的自我保护机制.
多个自噬相关基因及其单核苷酸多态性(single nucleotide polymorphism,SNP)与IBD有关,有研究证实IBD患者肠上皮细胞缺乏自噬使肠上皮细胞正常的生理平衡破坏.CD的风险位点ATG16L1[75]突变,表现出位于小肠隐窝底部的Paneth细胞自噬功能受损,影响细胞内物质和细胞器的降解与循环,减少抗菌物质的分泌以抵抗感染和清除胞内微生物[76,77],该缺陷被证明与患有CD风险增加有关,还会使DCs向T细胞呈递外源性抗原能力降低[78].此外,在全肠外营养大鼠模型中[79],无菌小鼠的肠上皮出现了自噬现象,当在补充丁酸盐后肠上皮恢复了完整性,提示可能大鼠结肠细胞中丁酸代谢受抑制,导致β-氧化、三羧酸循环和氧化磷酸化关键酶减少,ATP的合成受阻,AMPA激活,p27kip1磷酸化,最终导致肠道自噬.
微循环缺氧也是IBD的一个重要特征,在IBD患者肠道固有层和缺氧的肠腔中间,发现了“生理性缺氧”环境,该环境可诱导肠上皮细胞自噬以及抑制哺乳动物雷帕霉素靶蛋白/NOD样受体热蛋白结构域相关蛋白3(mammalian target of rapamycin/NOD-like receptor protein 3,mTOR/NLRP3)途径来调节机体炎症[80].其中,HIF-1α是维持氧稳态的转录激活因子[81],HIF-1α的上调可以抑制NF-κB信号通路发挥保护作用,减少细胞炎症,还能够影响肠道菌群的平衡,在维持肠道屏障稳态中起到了核心作用(如图2所示).丁酸通过诱导结肠细胞乏氧性代谢,增强胃肠道中HIF-1α的稳定性.通过FITC葡聚糖通量测量得出,丁酸盐可以促进T84细胞的屏障功能,而缺乏HIF-1α时可导致屏障功能受损.结果进一步提示,丁酸盐通过HIF-1α调节肠黏膜屏障功能[55].同时研究证明丁酸盐进入线粒体后,作为HDAC抑制剂刺激丙酮酸脱氢酶激酶(pyruvate dehydrogenase kinase,PDK)基因启动子,促进PDK基因表达,使丙酮酸脱氢酶复合物(pyruvate dehydrogenase complex,PDHC)活性降低[82],因此结肠细胞不能通过β-氧化将丙酮酸转化为乙酰CoA,抑制了氧化呼吸,导致生理性缺氧[55],该过程有利于维持HIF-1α的稳定性.HIF-1α可促进粘蛋白(mucins,MUC)分泌,使MUC2、MUC3[83]和ITF[84]上调,粘液层的厚度增加,为肠道微生物定植提供了空间.
环境缺氧能够降低CD患者结肠中的TNFα、Il-6和NLRP3的表达,诱导p62mRNA的表达和p62蛋白的减少,提示可能有自噬基因的翻转[80].有研究进一步证明HIF-1α和NF-κB参与调节黏膜缺氧反应在多个水平上密切相关.在低氧条件下,HT-29的细胞中HIF-1α与p62启动子结合增加,提示HIF-1α参与了自噬蛋白的表达;NF-κB参与了与NLRP3的启动子结合.这两种机制表明两个核因子的调控相反,与其拮抗功能一致[80].HIF-1α的稳定性增加可以抑制NF-κB,从而抑制促炎症反应.siRNA沉默HIF-1α后,发现Hela细胞IL-1β 和TNF-α等细胞因子诱导NF-κB产生,其活性显著增加[55].并伴随有NF-κB的特征性靶点p100和IL-8水平增加.同时,可见NF-κB靶基因A20、Cyld、DDX3、IAP1和XIAP的mRNA和蛋白质水平升高,虽然HIF-1α对NF-κB的抑制得到公认,但是,最新研究发现HIF-1α的过表达可降低对NF-κB抑制效应,推测该作用可能与机体的负反馈有关.综上所述,SCFAs通过调节肠道细胞的增殖与代谢相关蛋白的表达来影响肠道黏膜的完整性.
4 SCFAs保护肠道黏液屏障
肠道黏液屏障在肠道中起着润滑和物理屏障的双重作用,是肠道的第一道防线,主要由肠道Paneth细胞分泌的抗菌肽(antimicrobial peptides,AMP)和杯状细胞分泌的MUC和ITF组成,此外,还有免疫球蛋白A(immunoglobulin A,IgA)、活性转谷氨酰胺酶、热休克蛋白(heat shock proteins,HSPs)等.黏液除有润滑作用,还可减少酸和蛋白酶对肠道黏膜的侵蚀作用,同时也为肠道微生物提供适宜的生活环境.
不可否认SCFAs在肠道化学屏障的保护中具有重要的作用.AMP由IECs和Paneth细胞产生,能够抵御微生物群和病原体,AMP的不同家族包括防御素、杀微生物肽和C-型凝集素(如REG家族)[85].GPR43-/-的小鼠较WT小鼠肠上皮细胞的RegⅢγ和β-defensin 1、3、4表达水平均下降[86],口服SCFAs后,WT小鼠的RegⅢγ和β-防御素明显上升,而GPR43-/-的小鼠无变化,提示SCFAs通过GPR43依赖的方式促进肠上皮细胞生成AMP.此外,SCFAs能够激活mTOR和STAT3,用siRNA特异性敲除mTOR、用 siRNA特异性敲除STAT3并分别转染MSIE细胞[86],RegⅢγ和β-防御素的蛋白表达量均明显降低,说明丁酸可能通过激活mTOR和STAT3来调节AMP的产生.因而,SCFAs调节AMP产生具有GPR43依赖性,并且mTOR和STAT3对该途径具有一定的协同作用.
SCFAs在肠道物理屏障中作为调节器也发挥着不容小觑的作用,SCFAs可通过GPR43受体,促进肠上皮细胞分泌IL-18、抗菌肽、MUC,维持肠屏障的完整性.MUC的产生与muc基因有关,其中,muc1、muc3、muc4编码膜上的MUC,MUC2主要与肠道中分泌性MUC有关[87].MUC2是最受关注的一种黏蛋白,其改变与多种肠道疾病相关,它的寡聚糖链结构能为肠道菌群中的益生菌、IgA以及抗菌肽提供黏附位点,协助其定植,用以发挥生物屏障和免疫屏障的作用[88].有研究证实丁酸盐通过介导MUC2启动子处的AP-1和乙酰化/甲基化(acetylation/methylation),同时也参与MUC表达的调控中[89].使用丁酸盐和丙酸盐可诱导LS174T细胞MUC2 mRNA水平升高,该作用通过MUC2启动子内-947/-371处丁酸反应区,在这个区域中的-818/-808处发现了一个活性的AP1(c-Fos/c-Jun)顺式元件,介导丁酸诱导的启动子激活,同时发现丁酸盐对MUC2的调节与组蛋白H3和H4的乙酰化增加以及MUC2启动子H3的甲基化有关.至此可见,丁酸对MUC2的产生与MUC2基因的转录与表达具有一定的调控作用.在一项系统性研究中发现[90],UC患者MUC2结构或合成的改变对结肠黏膜完整性有影响,因而进一步的探索维持黏膜完整性是极其必要的.
肠黏液屏障是一个十分复杂的体系,SCFAs在IBD中的调节机制尚不明确,但不可否认的是,肠道黏液在IBD的发生发展中具有重要意义,而SCFAs在肠道黏液的调节中也是重要的一环.在未来的研究中,机制的完善或可以为IBD疾病的治疗提供新的靶点.
5 SCFAs加强肠道上皮细胞间的紧密连接
IECs依靠细胞旁路中的“看门人”维持上皮细胞的稳定性与组织间的稳态,这些“看门人”就是细胞间的粘附,包括紧密连接(tight junction,TJ)、黏附连接和桥粒[91].TJ和肠道粘液层是维持肠道完整的重要结构.其中,TJ位于相邻细胞之间,呈带状环绕细胞的顶部,由紧密连接蛋白Claudins、Occludins和封闭小带(zonula occludens,ZO)形成,可以在相邻细胞之间形成一道闭锁屏障,防止大分子物质进入,是机体防御病原体的重要机制.
5.1 SCFAs作用于紧密连接蛋白 TJ在控制细胞旁的通透性方面具有核心作用,TJ蛋白Claudins是一种具有高容量和选择性的通道,其中,Claudin-2对小的阳离子和水具有一定的渗透性,因此,Claudin-2导致的肠黏膜屏障损坏的患者可能会腹泻[92,93];ZO-1和occludin则与之相反,它们是一种低容量,非选择性的,只限制通过该通道的分子大小[94].SCFAs对紧密连接及其蛋白具有重要的调控意义,但是目前,SCFAs对于紧密连接蛋白的调控机制并未被完全阐明.
S C FA s能够修复屏障障碍,以T J为其作用靶点,比如Claudin-2是丁酸作用的主要靶点.有研究证实在Caco-2细胞中TNF-α、IFN-γ和白细胞介素13(interleukin-13,IL-13)添加到基底膜外侧,SCFAs添加至顶膜,作用24 h后[95],结果显示丁酸可以改善TNF-α/IFN-γ诱导的TER降低,并抑制TNF-α和IFN-γ介导的Claudin-2水平升高和Claudin-3水平降低,此外,丁酸盐还抑制了IL-13介导的TER降低和Claudin-2水平的升高,提示TNF-α/IFN-γ和丁酸均可以参与调节与肠黏膜屏障功能相关的AMP-活化蛋白激酶.TNF-α[96]和IL-13[97]通过PI3K通路使Claudin-2水平升高,该途径可以被丁酸抑制[98].PI3K通路还可以激活与细胞存活相关的Akt通路[99].由此,丁酸通过提高Claudin-2的含量以减轻肠屏障功能的障碍,有助于为肠黏膜屏障损坏相关疾病的治疗提供帮助.
突触足蛋白(synaptopodin,SYNPO)是另一种通过抑制HDAC受丁酸调控的肠上皮紧密连接蛋白,具有调节屏障功能和促进愈合的潜力[100].在T84细胞中发现SYNPO与ZO-1共定位于肠上皮细胞紧密连接处;SYNPO的抗体的定位具有非特异性,分布于非紧密连接的区域;应用phalloidin染色显示,用5 mM丁酸后与肌动蛋白弹力纤维相关的SYNPO的水平显著高于对照组[100].SYNPO通过调节肌动蛋白丝组装、应力纤维形成并维持细胞间相互作用参与肠上皮屏障功能[101].
有研究表明在DSS诱导的结肠炎时[100],SYNPO的蛋白表达量显著降低,并且结肠组织有炎性浸润和上皮隐窝损伤,而ZO-1的蛋白水平并无改变.但是TNF-α、IL-1β、INF-γ处理后的T84细胞中[100],可见SYNPOmRNA在中的表达降低.这些实验结果均说明了炎症对SYNPO的表达具有不利的影响.
与此同时,有研究证实丁酸盐通过抑制HDAC调节SYNPO,其对上皮修复功能通过肌动蛋白与SYNPO的偶联调控,而SYNPO是关键的微生物调节蛋白,它能够将肌动蛋白定位于紧密连接处,并形成一个动态的收缩以及信息交流中枢部位.SYNPO调节α-辅肌动蛋白4(α-actinin-4,ACTN4)的表达[102],并促进它在黏附连接处的累积,参与到细胞收缩的功能中,同时,丁酸可诱导ACTN4的表达[100].在T84细胞中,丁酸盐可以增加F-actin与总肌动蛋白的比值,但是,SYNPO-/-细胞中该比值没有增加.因此,丁酸需要肌动蛋白与SYNPO的偶联参与到上皮细胞伤口的愈合中.随着研究的深入,将发现更多紧密连接蛋白靶点,探索其作用机制,以期为IBD的治疗提供参考和理论基础.
5.2 SCFAs促进紧密连接的重组[103]在IBD患者的肠黏膜可见紧密连接的失调以及上皮细胞损伤.实验研究表明SCFAs可缓解小鼠结肠炎病理变化,其主要机制SCFAs通过激活腺苷酸活化蛋白激酶(adenosine 5’-monophosphate -activated protein kinase,AMPK),抑制(myosin light chain kinase/myosin light chain 2,MLCK/MLC2)通路,并介导(protein Kinase Cβ,PKCβ)磷酸化,使ZO-1、Occludin、Claudin-3和Claudin-4等肠上皮细胞连接蛋白的表达上调,促进紧密连接的形成[103].给予AMPK激动剂的小鼠溃疡性结肠炎较对照小鼠有明显改善,结肠组织中的炎性因子IL-18,IL-1β,COX-2,iNOS和IL-6的mRNA水平下降.肠组织发生损伤后,肠上皮紧密连接蛋白Claudin-1,Occludin和ZO-1的表达水平下降,肠上皮通透性增加,AMPK激动剂可以逆转这一变化[104].发现在活动性IBD患者中,MLC磷酸化水平显著增加,MLCK酶活性增加.使用Caco-2细胞模型,发现AMPK磷酸化水平升高可以导致Connexin43(Cx43)蛋白在丝氨酸368位发生磷酸化,Cx43与膜蛋白ZO-1结合降低并进入胞浆,使细胞膜上的ZO-1形成更为稳定的紧密连接,改善了肠上皮结构和功能.Miao等人发现[105],丁酸钠通过调节钙池诱导细胞外Ca2+内流(store-operated calcium entry,SOCE),激活Ca2+/CaMKKβ途径,从而激活AMPK.丁酸钠对紧密连接的重构通过MLCK/MLC2途径,其中丁酸钠可促进MLC2在Ser19处的磷酸化水平降低、PKCβ2的Ser660处或PKCβ1的Thr642处的磷酸化水平增加的过程(详细过程见图3).
以上结果表明,丁酸钠可介导A M P K磷酸化,进而抑制M L C2的磷酸化,促进P K C β2的磷酸化,p-Cx43(Ser368)水平升高,促进ZO-1形成紧密连接(如图3),并使结肠组织中的炎症因子转录水平下调,有利于Caco-2细胞中紧密连接的重组,有助于改善IBD.该机制也为IBD的治疗提供新的靶点和候选药物.
6 SCFAs影响肠黏膜屏障的通透性和水电解质的吸收
在对有CD家族史的家族进行队列分析,研究家族成员肠道通透性早期发病特征可能包括肠道通透性增加[6].肠上皮细胞具有高选择性的屏障的特点,这有助于小分子等营养物质从肠腔进入到黏膜下层.在Caco-2和HT-29细胞中,2 mmol/L丁酸诱导TER增高且通透性降低,而8 mmol/L的丁酸诱导通透性增加,Caco-2活细胞数量减少,表明丁酸调节肠上皮通透性与诱导细胞凋亡有关[58].不仅如此,TJ也会影响肠黏膜屏障的通透性.多个研究表明[106,107],在SCFAs参与到了水电解质的调节中,SCFAs的灌注家兔近端结肠显著减少结肠中水分的分泌,作用程度为: 丁酸>丙酸>乙酸.丁酸盐显著减少了Na+、Cl-和K+的分泌,但对HCO3-没有影响.SCFAs还可通过调节结肠上皮细胞内cAMP促进Na+的吸收[58,108].
7 总结
综上所述,SCFAs是肠道微生物代谢产物,亦是肠道健康的守护者.尤其是丁酸盐,作为肠道菌群和肠道黏膜屏障之间的桥梁受到广泛关注.SCFAs通过多种机制调节黏膜屏障作用,重构黏膜屏障,缓解IBD症状.目前,一些新型的生物制剂和小分子药物也在IBD的治疗中展现出了一定的潜力,医生对IBD的治疗也逐渐趋向于个体化,即将迎来更多的选择与挑战.对SCFAs的深入研究或可成为治疗相关疾病的一个研究方向.
