Alg13突变致痫小鼠的繁育及鉴定
2022-11-25霍俊明郭宝瑞于宝利王军成
高 鹏,霍俊明,郭宝瑞,于宝利,吴 际,王军成,马 辉,孙 涛,张 静
750004 银川,宁夏医科大学总医院:神经外科1,外科学研究室8;750004 银川,宁夏医科大学:颅脑疾病重点实验室2,生育力保持教育部重点实验室6;014010 内蒙古 包头,内蒙古科技大学包头医学院第一附属医院心血管外科3;215400 江苏 太仓,依科赛生物科技(太仓)有限公司4;200030 上海,上海交通大学Bio-X研究院遗传发育与精神神经疾病教育部重点实验室5;750002 银川,宁夏回族自治区人民医院神经外科7
ALG13(UDP-N-acetylglucosaminyl transferase subunit)是X连锁的基因,编码ALG13蛋白,其与ALG14形成尿苷二磷酸-N-乙酰葡萄糖胺转移酶(UDP-N-acetylglucosamine transferase)催化N连接的糖基化[1]。ALG13蛋白天冬酰胺N连接糖基化是蛋白质修饰中最常见和普遍的形式,糖基化过程的紊乱会引发多种疾病[2-3]。2012 年,研究人员首次发现一名患有ALG13缺乏症的男孩,该男孩具有的c.280A>G突变导致其出现耐药性癫痫发作、小头畸形、眼球震颤、双侧视神经萎缩、锥体外系体征、肝肿大、水肿和出血倾向,并在1 岁时死亡[4]。之后,一系列对婴儿痉挛症、Lennox-Gastaut 综合征、West 综合征等伴发严重癫痫和智力障碍疾病患者的筛查发现ALG13中的新生突变(如:c.320A>G)与疾病的发生发展密切相关[5-7]。因此,X 连锁ALG13基因变异可能是癫痫性脑病的潜在原因。
癫痫是一种以脑神经元过度异常放电且反复发作为特征的异质性疾病。虽然许多病理生理的变化会影响癫痫易感性,但研究表明遗传是癫痫疾病发生发展的重要因素[8]。与癫痫症状相关的基因突变可能会使配体或电压门控离子通道异常,其中已广泛研究确认的癫痫易感基因SCN1A——编码 Nav 1.1 钠通道的 α 亚基,该基因的异常表达导致严重的遗传性癫痫综合征[9-10]。糖基化是维持钠通道生物合成和降解的正常稳态的必要过程,ALG13突变造成的糖基化紊乱可能会使钠通道结构和功能异常,进而导致癫痫的发生。
小鼠癫痫模型可分为诱发性癫痫模型和遗传性癫痫模型。诱发性癫痫模型是通过化学、电或声刺激导致急性或慢性癫痫发作;遗传性癫痫模型则通过遗传病变(或自发突变)导致自发性癫痫发作,更能有效反映某种特异性的癫痫。目前,Alg13基因缺陷型致痫小鼠模型尚未见报道。因此,本研究对Alg13基因敲除小鼠进行了繁育和鉴定,并对其方法学进行探讨和优化,确保该模型能够成为Alg13缺陷所致癫痫研究的良好动物模型。围绕 ALG13活性水平,开展其在癫痫中的作用及机制研究,可能为理解癫痫的发病和治疗提供一个新的视角。
1 材料与方法
1.1 材料
1.1.1 实验动物 C57BL/6J背景的Alg13KO小鼠由于宝利博士友情惠赠[11]。本实验选用4周龄Alg13KO小鼠12只(雌雄各6只;体质量6~7 g),野生型(wildtype, WT)小鼠12只(雌雄各6只;体质量7~8 g),8周龄Alg13KO雄性小鼠18只(体质量21~23 g),WT雄性小鼠18只(体质量22~24 g),分为Alg13KO组和WT组。本实验遵循宁夏医科大学实验动物管理和使用委员会的规定[SCXK(Ning)2015-0001]。
1.1.2 试剂与仪器
1.1.2.1 主要实验试剂 本实验采用的主要实验试剂有:琼脂糖(美国Pharmacia 公司),DNA提取试剂盒(北京天根生化科技有限公司),DNA产物纯化试剂盒(北京天根生化科技有限公司),TaqDNA聚合酶(美国Promega公司),T4 连接酶(美国赛默飞有限公司),T4连接酶缓冲液(美国赛默飞有限公司),pCRTM4-TOPO® TA载体(美国赛默飞有限公司),异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactoside, IPTG)和5-溴-4-氯-3-吲哚基-β-D-半乳糖苷(5-bromo-4-chloro-3-indolyl-β-D-galactoside,X-gal)(北京索莱宝科技有限公司),免疫组化染色试剂盒(北京中杉金桥生物技术有限公司),ALG13抗体(武汉三鹰生物技术有限公司),SCN1A抗体(美国Abcam公司),SDS-PAGE凝胶快速配制试剂盒(碧云天生物技术有限公司),全蛋白提取试剂盒(南京凯基生物科技发展有限公司),BCA蛋白浓度测定试剂盒(南京凯基生物科技发展有限公司),蛋白Marker(美国赛默飞有限公司),ECL发光液(美国赛默飞有限公司),山羊血清封闭液(北京中杉金桥生物技术有限公司)。
1.1.2.2 主要仪器设备 分光光度计(美国赛默飞有限公司),ABI基因扩增仪(美国应用生物系统公司),BIO-RAD Gel Doc XR+凝胶成像系统(美国伯乐公司),DYY-7C电泳仪(北京市六一仪器厂),Lecia RM2265石蜡切片机(德国Lecia公司),Leica DM6正置双目生物显微镜(德国Lecia公司)。
1.2 方法
1.2.1Alg13基因敲除小鼠的饲养 将动物饲养在宁夏医科大学SPF级实验动物中心独立通气笼盒(individually ventilated cages, IVC)设备系统中,维持在具有恒定室温(24±0.5)℃,相对湿度50%~60%,自由饮用食物和水,12 h/12 h光/暗循环中,每笼饲养动物数量不超过14只。使用SPF级大小鼠繁殖饲料、高压灭菌垫料及超纯水,每日为鼠笼补充饲料及饮水,每周更换垫料2次,每2周更换水瓶1次。
1.2.2Alg13基因敲除小鼠的繁育 先将Alg13KO的雄性纯合子(Alg13X-Y)与雌性杂合子(Alg13X+X-)按1∶2合笼,繁育出子代小鼠后进行基因型鉴定,挑选出WT雄鼠(X+Y)、雌鼠(X+X+)、KO雄鼠(X-Y)和雌鼠(X-X-),分别对WT和KO小鼠进行繁育,每笼雌鼠生育3代后停止此笼繁育,选用其子代小鼠重新合笼繁育。
1.2.3Alg13基因敲除小鼠的鉴定
1.2.3.1 小鼠尾部DNA的提取及PCR扩增反应 待子鼠4周龄时,剪取小鼠尾部组织加入490 μL的裂解缓冲液和10 μL 蛋白酶K,60 ℃ 水浴过夜。使用DNA提取试剂盒,按照试剂说明书提取小鼠基因组DNA,应用分光光度计测定其浓度。引物序列按照Alg13的第4外显子序列设计正、反向引物,正向:5′-CAAATGGGTAACAGGC-3′;反向:5′-GGGTAGAAAA-GGATGG-3′。对于每个样品,反应体系为:5 μL正、反向引物,12 μL无核酸酶水,25 μL Q5聚合酶和8 μL(60 ng/μL)DNA。在ABI PCR基因扩增仪上设置反应条件:98 ℃下保持30 s后(预变性),进行35个PCR循环[98 ℃10 s(变性),56 ℃ 30 s(退火),72 ℃ 2 min(延伸)],然后在72 ℃ 2 min(延伸)后终止反应。
1.2.3.2 琼脂糖电泳法 取PCR扩增产物5 μL加入1 μL的6×DNA 上样缓冲液在2%琼脂糖凝胶中以90 V由负极向正级电泳30 min,使用100 bp的DNA Ladder作为DNA标记。于化学发光凝胶成像系统中观察结果,并将得到的PCR产物进行测序,从而获得基因型结果。
1.2.3.3 TA克隆法 PCR产物在琼脂糖凝胶电泳后,紫外灯下切除目的条带周边多余凝胶,对DNA进行纯化。将20 μL纯化产物加入含有1 μLTaq聚合酶,1 μL dATP和8 μL ddH2O的反应体系中,在72 ℃的水浴中孵育30 min。将1 μL T4连接酶,1 μL T4连接酶缓冲液,5 μL已加过‘dA’的纯化DNA,2 μL ddH2O,1 μL pCRTM4-TOPO®TA 载体共同加入体积为200 μL无酶管中,于16 ℃的水浴中过夜连接。上述克隆溶液取5 μL加入到含有50 μL感受态细胞悬浮液的1.5 mL无菌EP管中,混匀后置于冰浴中30 min。随后将500 μL LB培养基加入EP管中并在37 ℃恒温摇床下振荡1 h,将振荡好的菌液进行短暂的离心,弃去300 μL上清液,涂抹在含有IPTG和X-gal以及氨苄青霉素的固体培养基上,在37 ℃的恒温培养箱中倒置培养14 h。观察培养基中蓝色与白色菌落形成数量与密度,从菌落中选取20个白色菌落,分别放入20个含有氨苄青霉素的LB培养基中,将摇菌管放于37 ℃恒温摇床中培养16 h;提取质粒并进行DNA测序。
1.2.4Alg13基因敲除小鼠的ALG13蛋白表达检测
1.2.4.1 免疫组化分析 小鼠腹腔注射1%戊巴比妥钠麻醉,经心脏灌注并用4%多聚甲醛进行快速固定。断头后迅速取出脑,4%多聚甲醛固定,经梯度浓度酒精中逐级脱水,二甲苯透明处理后将脑组织浸蜡包埋,使用石蜡切片机切片。将石蜡切片放入二甲苯脱蜡、梯度酒精中水化后,用5%正常山羊血清封闭1 h,然后在4 ℃下与兔抗ALG13抗体(1∶300)孵育过夜。洗涤后将切片与生物素化的山羊抗兔IgG在室温下孵育1 h。DAB显色,显微镜下观察组织的显色情况,发现有棕黄色阳性表达可用自来水终止反应。苏木精染色、1%的盐酸酒精分化、梯度酒精脱水、二甲苯透明处理,中性树胶封片后置于显微镜下进行拍照,组织切片以出现棕黄色或褐色颗粒可认为是阳性表达。应用Image-Pro Plus 6.0软件,计算其平均光密度(IOD/area)。
1.2.4.2 Western blot分析 处死小鼠取脑后,分离出约50 mg海马、大脑皮质和小脑,提取蛋白并测定样本蛋白质浓度。电泳时的参数设置为:浓缩胶(80 V,30 min),分离胶(100 V,90 min)。将含有目的蛋白的分离胶块在低温环境下300 mA转膜1 h。经过5%的封闭液封闭1 h后,加入一抗并于4 ℃冰箱过夜孵育。洗膜3次后,与二抗孵育1 h。采用ECL发光法得到X线片用Bio-Rad凝胶成像系统进行拍照,应用Image J软件测定目的蛋白的表达情况。
1.2.5 小鼠颅骨外植入视频脑电图(video-electroencephalography, VEEG)设备和脑电记录 小鼠腹腔注射1%戊巴比妥钠麻醉后置于立体定位框架上,备皮处用酒精消毒并沿中线用手术刀切开暴露头骨。以前囟为原点,前囟向后 1.5 mm,正中线左右旁开 2 mm,将自制电极固定颅骨,置于左额叶皮层的电极为参考电极,右顶叶的电极为记录电极,用牙科水泥进行固定,待麻醉恢复后将小鼠单笼饲养。1周后将记录系统与小鼠头部的电极连接,将视频监测和脑电记录同步,以便同时监测小鼠的行为学改变。设置好参数,待系统稳定后开始记录,每次记录24 h,连续记录14 d。SleepSign 信号采集系统记录小鼠皮层脑电活动,并用 SleepSign和Matlab对脑电信号傅里叶转换及频谱分析。
1.2.6 癫痫发作严重程度评价 小鼠的癫痫发作情况依据Racine评分来进行评估和分类,0级:正常行为,无癫痫发作;1级:口腔和面部运动,表现为面部抽动,包括眨眼和胡须抖动;2级:颈部肌肉痉挛或出现有节律性的点头或者甩头;3级:出现单侧前肢阵挛或双侧前肢阵挛;4级:双侧前肢阵挛伴双后肢站立;5级:全面性的强直阵挛发作伴失去平衡摔倒。
1.3 统计学分析
2 结果
2.1 Alg13 KO小鼠的外形及基因型的鉴定
C57BL/6遗传背景的Alg13KO小鼠成年为黑色,与野生型无差异。繁殖小鼠交配后的子代新生仔鼠与野生型仔鼠外观形态无显著性差异。Alg13KO小鼠通常表现出发育迟缓,但存活下来的Alg13KO小鼠在成年时与WT小鼠体型接近(表1)。
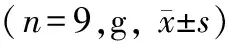
表1 WT组和Alg13 KO组小鼠出生后体质量比较
通过PCR扩增含有“TTCAT”的DNA片段,由于“TTCAT”序列的删除,X+Y和X+X+小鼠预期的PCR产物为545 bp,而X-Y和X-X-小鼠预期的PCR产物为异常的540 bp,对于杂合X+X-小鼠,存在540 bp和545 bp两种产物(图1A)。因琼脂糖凝胶电泳产生两个条带难以区分,故对小鼠PCR产物进行了DNA测序。结果显示Alg13KO小鼠中存在“TTCAT”缺失,但WT小鼠中保留了“TTCAT”,杂合小鼠在“TTCAT”缺失位置显示出双色重叠峰(图1B)。为了得到量大而且序列单一准确的目的片段,对PCR产物进行TA克隆和蓝白斑筛选,含有重组DNA的靶向载体(图1C)转化的细胞产生白色菌落,非重组质粒转化的细胞会长成蓝色菌落(图1D)。取白色菌落测序分析显示WT小鼠序列的341至345位含有“ATGAA”5个碱基,即对应目的基因上序列的225~229位“TTCAT”,Alg13KO小鼠缺少这5个碱基,而杂合型则含有两种类型的序列(图1E、F)。以上结果表明本研究保证了Alg13基因敲除小鼠的种群延续和遗传稳定性。
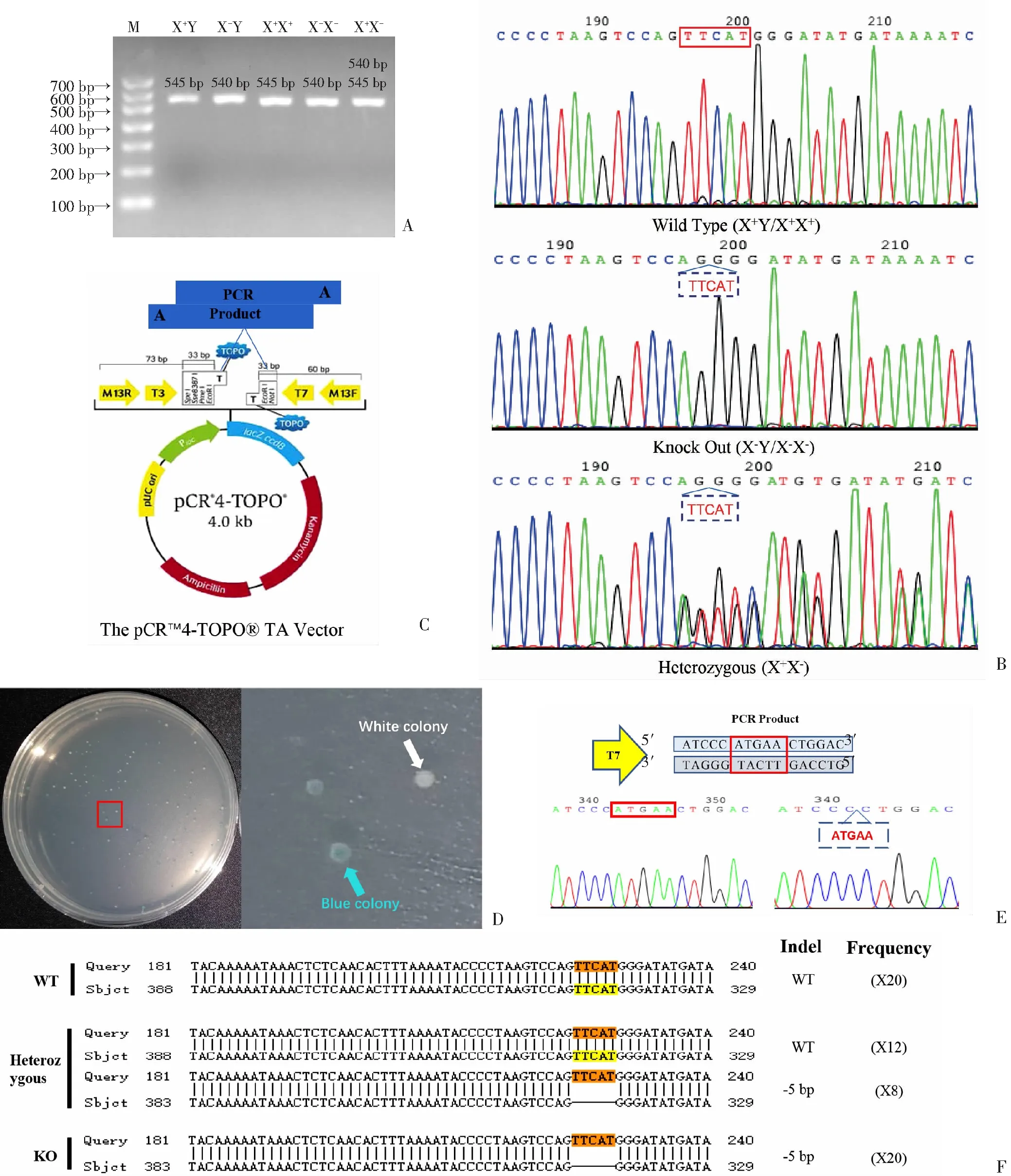
A: WT、Alg13 KO和杂合子小鼠PCR产物的2%琼脂糖凝胶电泳分离结果 WT小鼠显示545 bp的片段,Alg13 KO小鼠显示540 bp的片段,杂合子小鼠显示540 bp和545 bp两种片段 M:示DNA标记物;B:正向引物和反向引物扩增WT、Alg13 KO和杂合子小鼠PCR产物的DNA测序结果 红色实线矩形:示“TTCAT”,蓝色虚线矩形:示敲除的“TTCAT”;C:目的基因靶向载体结构示意图;D:WT、Alg13 KO和杂合子小鼠目的基因蓝白斑筛选 红色矩形:示放大图像范围,蓝色箭头:示蓝色菌落,白色箭头:示白色菌落;E:WT、Alg13 KO和杂合子小鼠目的基因白色菌落质粒提取物的DNA测序 红色实线矩形:示“ATGAA”,蓝色虚线矩形:示敲除的“ATGAA”;F:WT、Alg13 KO和杂合子小鼠NCBI核苷酸BLAST结果分析 橙色:示原始Alg13基因,黄色:示保留的“TTCAT”
2.2 Alg13KO小鼠ALG13蛋白的表达
采用免疫组织化学法和Western blot评估基因的缺失对小鼠脑中ALG13蛋白表达的影响。对大脑皮质、小脑和海马进行免疫组织化学分析,结果显示:WT和Alg13KO同窝小鼠在脑组织中ALG13蛋白表达具有明显差异。在出生后8周,WT小鼠6个皮质层中观察到抗ALG13抗体孵育后神经元的强烈细胞质免疫反应性;在Alg13KO小鼠的相同位置未观察到明显的阳性神经元(P<0.001,图2A、B)。对于WT小鼠,细胞质的小脑颗粒层中显示有ALG13标记的神经元,并且大多数浦肯野细胞出现细胞质免疫反应性,而Alg13KO小鼠为ALG13阴性(P<0.05,图2A、B)。类似地,WT小鼠的海马CA1区中的多个神经元显示出对ALG13的强细胞质免疫反应性,而Alg13KO小鼠中观察到相同位置ALG13阳性神经元的显著缺失(P<0.001,图2A、B)。Western blot检测结果显示:Alg13KO小鼠缺乏对应于ALG13的条带,定量分析也表明Alg13KO小鼠的ALG13蛋白表达量显著低于WT小鼠(P<0.001,图2C、D)。
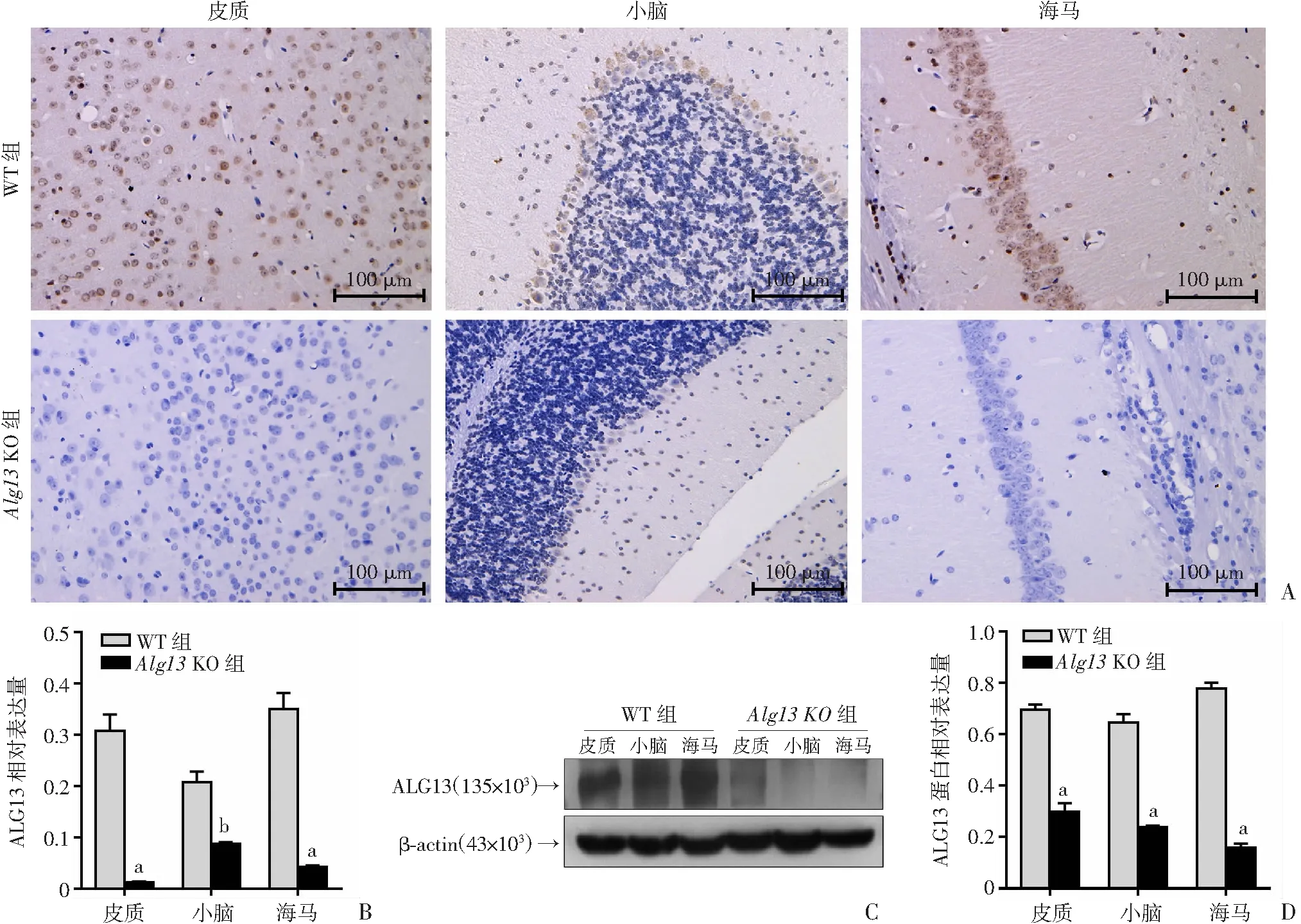
A:免疫组化检测小鼠皮质、小脑、海马中ALG13的表达;B:免疫组化定量分析;C:Western blot 检测小鼠皮质、小脑和海马区域ALG13蛋白表达;D:半定量分析 a: P<0.001, b: P<0.05,与WT组比较
2.3 ALG13缺乏导致自发性癫痫发作
出生后2个月,Alg13KO小鼠在换笼时偶尔表现出癫痫发作。通常,癫痫发作的行为表现始于颈部和尾部的张力过高,其次是重复的前肢阵挛,伴有双下肢站立并失去的姿势,在发作后期观察到会有短暂的静止状态(图3A)。利用同步的视频脑电系统长期监测小鼠脑电表现(表2),发现Alg13KO小鼠脑电显示出明显异常,典型特点如下:①发作前期主要是低幅高频的电活动;②发作后期主要为高幅低频尖波爆发(图3C)。然而,对于行为学上未表现出癫痫发作的Alg13KO小鼠,仍可记录到频繁地被定义为“EEG 癫痫”的脑电活动,具体表现为持续时间大于5 s,波幅大于2倍基线,频率大于 5 Hz的电活动(图3B)。而这种异常脑电活动从未在野生型小鼠中被监测到(图3C)。长期记录Alg13KO小鼠和 WT小鼠的基础脑电活动,为更准确地分析,脑电频段被规定为:δ(0.25~4 Hz),θ(4~12 Hz),α(12~18 Hz),β(18~30 Hz)和低γ(30~50 Hz)。快速傅里叶转换分析结果显示,较之野生型小鼠,Alg13KO小鼠脑电δ和θ波段能量值显著升高(P<0.001),频谱分析结果同样显示Alg13KO小鼠脑电δ和θ波段能量值显著升高(P<0.001,图3D~F)。
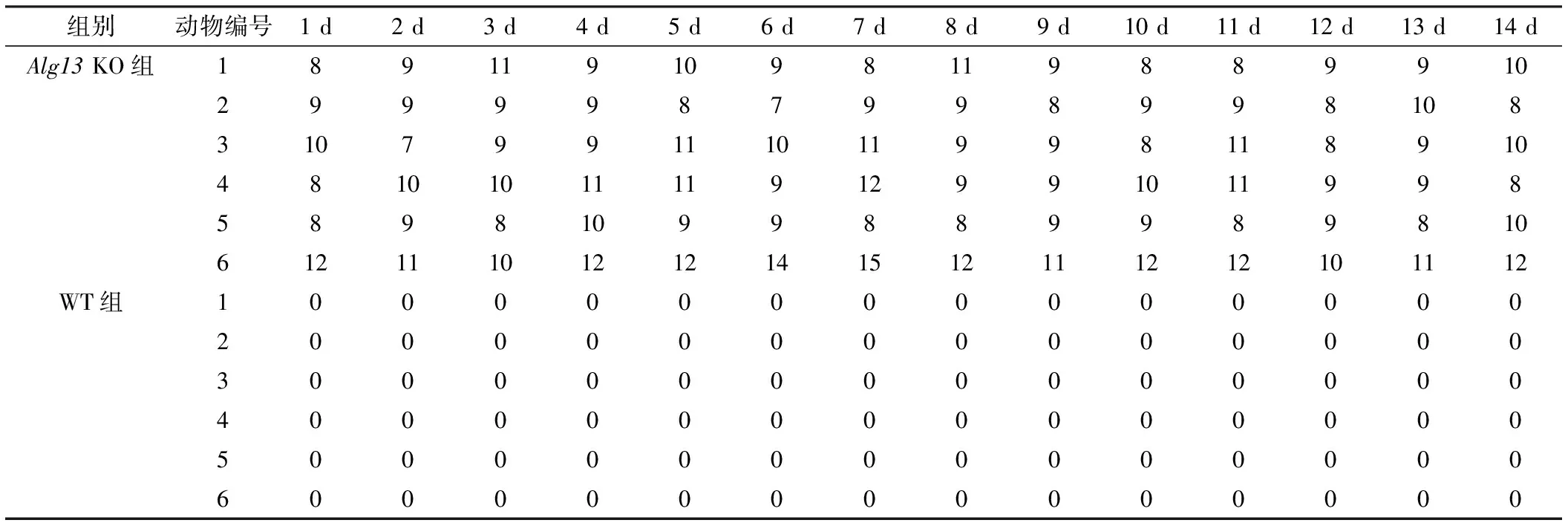
表2 模型小鼠自发性“EEG癫痫”事件的频次 (次,n=6)
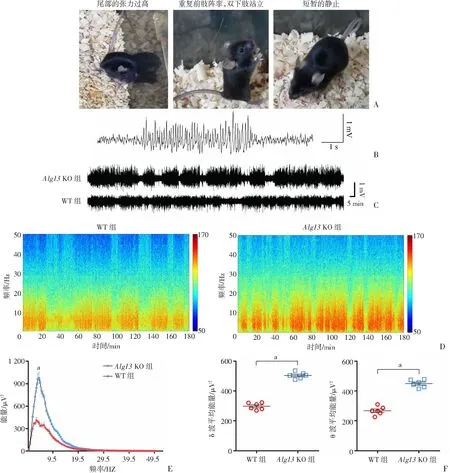
A: Alg13 KO小鼠自发性癫痫发作症状观察;B:“EEG 癫痫”的示例 被定义为出现高振幅(>2倍线)、高频率(>5 Hz)、持续 5 s以上的放电;C:Alg13 KO小鼠和WT小鼠脑电活动轨迹图;D:Alg13 KO小鼠和WT小鼠代表性脑电图分析的光谱图;E:两组小鼠脑电图数据的傅里叶转换分析;F:脑电δ和θ波的平均能量值比较 a: P<0.001
2.4 Alg13 KO小鼠模型中SCN1A的表达变化
为了评估由ALG13缺乏引起的癫痫发作是否与钠通道表达的变化相关,提取来自小鼠皮质和海马的细胞膜蛋白,并通过Western blot检测SCN1A的表达。结果显示:Alg13KO小鼠的海马和皮质中SCN1A的表达水平显著高于WT小鼠(图4)。
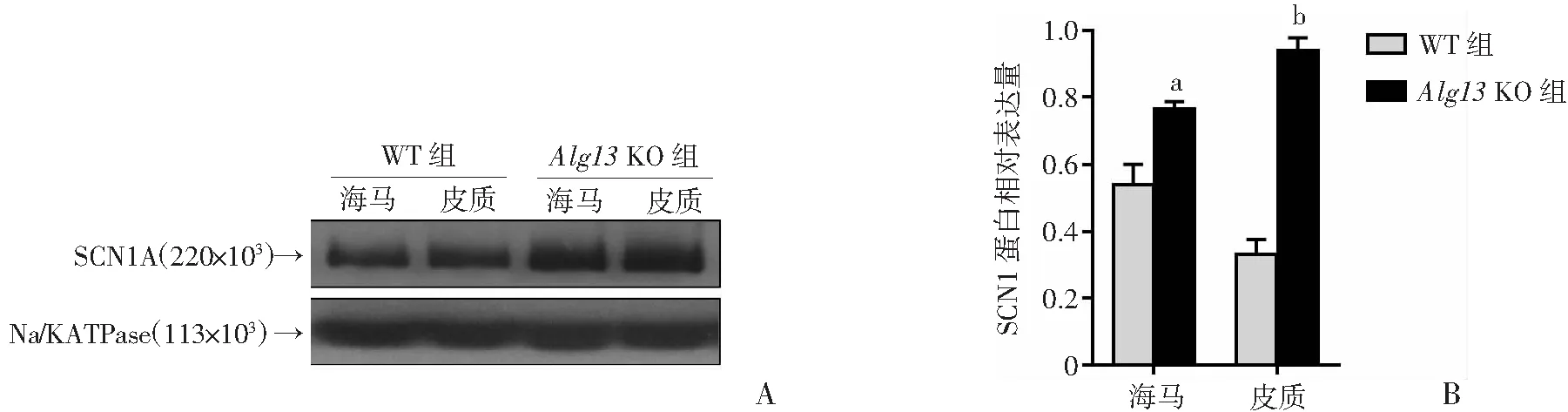
a: P<0.05,b: P<0.01, 与WT组比较
3 讨论
3.1 Alg13基因敲除小鼠是癫痫研究的良好动物模型
动物模型已广泛应用于包括癫痫在内的人类神经系统疾病研究中。尽管人类与小鼠在神经系统的结构、组成和功能各方面都存在差异[12],但由于人类与小鼠的基因具有超过99%的同源性,且基因具有较强的时空表达保守性[13],小鼠仍是研究人类神经疾病的理想动物模型之一。就癫痫而言,在人类和小鼠模型中,皮质和海马结构都是癫痫发作的主要位置[14]。小鼠癫痫模型可分为诱导模型和遗传模型。在诱导模型中,化学、电或声刺激会导致急性或慢性癫痫发作,具体取决于诱导方法和方案。在遗传模型中,遗传损伤(或自发突变)导致自发性癫痫发作的出现[15]。本研究对CRISPR-Cas9基因编辑技术构建的Alg13基因敲除小鼠进行了基因型鉴定、ALG13蛋白表达检测、癫痫发作情况的分析以及癫痫相关基因表达的研究,该模型表现出显著的癫痫易感性,为深入研究ALG13在癫痫中的作用提供一种理想的敲基因动物模型。
基因鉴定是基因敲除小鼠保种繁殖的重要环节,其中常见小鼠基因敲除鉴定所使用的检测方法之一为 PCR法。本研究采用小鼠合笼法获得子代,并提取鼠尾组织基因组 DNA,利用PCR扩增和凝胶电泳等鉴定Alg13敲除鼠的基因型,但由于敲除的Alg13基因的碱基数量较小,所以在电泳图中观察不到显著差异。因此对该基因片段进行测序验证,确保相应的碱基被成功敲除。本研究还应用蓝白斑筛选联合测序技术,对Alg13基因的缺失突变进行检测。蓝白斑筛选技术同时具有高效性和灵敏性,利用蓝白斑技术的高效性可以克服一代测序技术对稀有突变检测低的灵敏度。通过蓝白斑筛选、酶切与一代测序相结合对突变进行分析,结果显示,此方法可以成功检测Alg13基因的缺失。通过免疫组化和 Western blot检测小鼠大脑皮质、小脑和海马区中的 ALG13蛋白,结果也证实该基因被成功敲除。电压门控离子通道对于神经元兴奋性是必要的,并在癫痫发生中起关键作用[16]。电压敏感性钠通道是由大的中心成孔糖基化α亚基和两个较小β亚基组成的异聚复合物,其中大多数功能依赖于糖基化的α亚基。α亚基的N-糖基化对于钠通道生物合成是必需的,并且已知ALG13参与UDP-GlcNAc糖基转移酶的形成以催化蛋白质的N-糖基化[17]。因此,本研究对ALG13与一个重要的癫痫发生相关基因SCN1A的关系进行了分析。结果显示,缺乏ALG13可能影响Nav 1.1在可兴奋细胞膜中发挥功能的正常N-糖基化,进一步导致SCN1A的异常表达,间接引起自发性癫痫发作和癫痫易感性增加。以上结果表明,本研究构建的小鼠能够成为后续研究的良好动物模型。
3.2 Alg13基因异常对脑发育和脑功能的影响可能是癫痫产生的潜在机制之一
ALG13相关的先天性糖基化障碍(ALG13-CDG),也被称为早期婴儿癫痫性脑病-36(early infantile epileptic encephalopathy 36, EIEE36),是由位于Xq23染色体上的Alg13基因的杂合突变引起的。ALG13-CDG的临床表现包括癫痫、发育延迟、智力障碍、肌张力低下、凝血异常和内分泌功能障碍等[18]。ALG13编码一种与ALG14异源二聚化的蛋白,在内质网中形成功能性UDP-GlcNAc糖基转移酶[6, 19]。蛋白天冬酰胺的N-糖基化对于糖蛋白的结构和功能是必不可少的,因为它能催化蛋白N-糖基化的第二步,这也是患者表现出可变的多系统表型的原因。ALG13基因突变被认为是I型糖基化异常所致先天性疾病的原因,并且在癫痫性脑病的病因学中起作用。本研究发现,ALG13缺失与小鼠癫痫之间可能存在关联[20]。人类和小鼠之间存在遗传上的高度相似性和在神经生理学上较小程度的相似性[21]。研究中所建立的Alg13敲基因小鼠癫痫模型,已经在转录组学、神经成像、神经生理学和行为学的研究中应用,结果显示出某种形式的癫痫易感性和神经发育异常,但没有明显的N-糖基化缺陷表现[20]。因此,这一模型尚缺乏作为儿童发育性和癫痫性脑病(developmental and epileptic encephalopathy, DEE)直接模型的证据支撑,但仍可作为新生儿因发育异常所致癫痫症状的研究模型。此外,ALG13缺失在涉及癫痫相关小鼠脑发育、功能活动和信号通路的蛋白质翻译后糖基化修饰中的作用知之甚少[22]。需进一步研究以分析小鼠N-糖基化的潜在变化以及它引起癫痫的可能机制。
ALG13调控的N-连接糖基化对脑神经元发育至关重要。在大脑中,神经元、少突胶质细胞和星形胶质细胞是从神经干细胞发育而来的,源自神经干细胞的细胞依赖于通过糖基化与其他细胞和分子相互作用[23]。糖基化蛋白参与神经元和胶质细胞中 γ-氨基丁酸受体A[γ-aminobutyric acid receptor A,GABA(A)Rs]和水通道蛋白 4(aquaporin 4, AQP4)蛋白复合物的组装,在发育过程中,糖基化缺陷会损害其与细胞外基质(extracellular matrix, ECM)相互作用的能力,常导致脑发育的异常[24]。此外,在几乎所有参与调节或构成神经元信号传导的分子过程的蛋白质上都能观察到 N-连接聚糖,虽然并非所有的 N 连接聚糖修饰对于正确的神经元信号传导都是至关重要的,但其中很大一部分能够显著影响神经元活动[3]。例如,电压门控离子通道(voltage-gated ion channel, VGIC)上的唾液酸化 N-连接聚糖是VGIC 活性最有效的调节剂之一,添加N-连接聚糖会显著增加轴突放电的速率[25]。位于突触小泡中的几种质子泵和神经递质反转运蛋白也是糖蛋白[26]。因此,N-连接聚糖将新陈代谢与神经元内的信号传导相结合,正确控制 N-连接糖基化对神经元功能至关重要。癫痫正是一种由脑神经元异常放电引起的慢性脑病,ALG13基因的突变可能通过影响参与脑发育以及神经传导过程的各类蛋白的糖基化从而导致癫痫的发生。
3.3 研究的局限性和总结
本研究建立的癫痫小鼠模型中Alg13的基因突变是缺失突变,而一些癫痫性脑病患者一般为点突变(如c.320 A>G)[27-28]。基因突变类型的这种差异可能对ALG13蛋白的功能产生不同程度的影响。虽然本研究结果表明Alg13缺失突变导致小鼠癫痫发作,但要完全解释ALG13在人类癫痫发病机制中的作用还需进一步的研究。
综上所述,本研究成功建立了Alg13基因敲除小鼠模型,该模型表现出明显的自发性癫痫发作和癫痫易感性,可用于遗传异常导致癫痫的机制研究。
利益冲突声明文章内容不涉及相关利益冲突,该研究未涉及任何厂家、相关雇主或其他经济组织直接或间接的经济或利益的赞助
