基于深度学习的“药物-靶点”亲和力预测方法设计
2022-11-18李添添王俊杰
李添添,王俊杰
药物发现是从化学数据库中为特定的疾病寻找新药并在临床试验中验证其有效性和安全性的过程。这个过程通常需要10 年以上,期间消耗大量的时间和人力成本,但其结果往往不尽人意[1]。事实上,根据汤森路透生命科学事业部一份报告,2008-2010 年,108 种新的或重新利用的药物中,有51%由于疗效不佳而被宣告为失败[2]。这份报告提出以下两点在药物研发过程中十分重要:一是选择新的及更合适的药物靶点;二是在药物研发的最初阶段,尽可能筛选出疗效良好的药物。因此,预测药物和靶点之间的相互作用具有重大意义。
然而长期以来,“药物-靶点”相互作用的预测被认为是一个简单的二分类问题[3-4],即仅预测药物与靶点之间是否存在相互作用,而很少对它们关系的亲和力值进行评价。亲和力值可以提供药物与靶点相互作用的强度信息,能够对候选药物做出更为全面的评价[5]。目前在“药物-靶点”亲和力的预测任务中,Kronecker 正则化最小二乘[6](Kronecker regularized least squares,KronRLS)是一种基于相似度的方法,即采用不同类型的药物相似度和蛋白质相似度评分矩阵作为特征,将“药物-靶点”亲和力的预测问题表述为一个回归或秩预测问题;SimBoost[7]是一种新颖的使用梯度增强回归树的非线性方法,该方法同样使用相似的矩阵和构造特征,其训练数据的定义类似于KronRLS 方法。这两种方法均是基于特征工程的传统机器学习方法,但其预测结果的准确率仍不尽人意。得益于深度学习在图像处理和语音识别的成功应用[8],深度学习方法也被广泛应用于生物信息学领域,如基因组学研究[9]和药物发现[10]。深度学习的主要优势在于通过在每一层的神经网络中进行非线性转换,可以更好地表示原始数据,从而有助于学习数据中隐藏的模式[11]。Öztürk Hakime 等[12]在2018年首次提出使用深度药物-靶点亲和力(deep drug-target binding affinity,DeepDTA)的方法预测“药物-靶点”亲和力。该方法使用药物化学结构的一维表示作为药物的输入数据,氨基酸序列用于表示靶蛋白的输入数据。但是在该方法中,氨基酸序列使用的独热编码方式仅独立地描述了每一种氨基酸,并没有考虑肽链的上下游信息,也无法突出哪些氨基酸对靶蛋白有重要的修饰作用。因此,本文将改良以上DeepDTA 方法,构建一种准确率更高的基于深度神经网络的“药物-靶点”亲和力预测方法。
1 研究方法
研究方法概述如下:先对拟定药物进行独热编码,再通过双向长短时记忆网络[13-14](bidirectional long short-term memory,biLSTM)预训练语言模型对蛋白质(氨基酸序列)进行编码,随后将药物的独热编码和蛋白质的编码通过预测网络模块进行深度学习,得出二者的相互作用分数,最后将预测结果在Davis 激酶结合亲和力数据集[15]和KIBA大规模激酶抑制剂生物活性数据集[7]上进行验证。方法框架见图1。
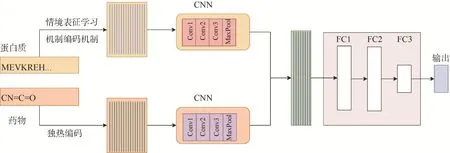
图1 研究方法框架
1.1 药物(化合物)的独热编码机制
从PubChem BioAssay 数据库中收集200 万个结构多样的化合物的简化分子线性输入规范(simplified molecular input line entry system,SMILES)序列,筛选出64 个描述符,每一个描述符对应特定的整数作为SMLIES 的独热编码,如字符“C”对应整数1,“N”对应整数3,“O”对应整数5,“=”对应整数63,则SMILES“CN=C=O”的独热编码为向量[1,3,63,1,63,5]。
1.2 双向LSTM 预训练氨基酸编码机制
使用预先训练的多层BiLSTM 获得氨基酸序列的向量表征。首先,对一条氨基酸序列(r1,r2,…,rN),biLSTM 语言模型分别使用M个堆叠的LSTM 网络从前向和后向2 个方向计算氨基酸出现的概率,2 个方向的LSTM 分别基于前向和后向语言模型的上下文输出中间嵌入向量(即隐藏状态向量),其中j=1,…,M。再对每一个氨基酸ri使用M层的双向语言模型计算出2M+1 个嵌入向量E(ri)={hij|j=0,…,M}。然后通过聚合不同层的表示获得其上下游的信息表示。因此,一条氨基酸序列(r1,r2,…,rN)经过双向语言模型编码后表示为一组等长的向量Eco(S)=[Eco(r1),Eco(r2),…,Eco(rN)]。设定M=2,即使用2 组双向LSTM 编码氨基酸序列,将其中每个LSTM 的隐藏单元设定为32。BiLSTM 预训练模型的网络结构如图2 所示。为了充分预训练BiLSTM 模型,从STRING 数据库[16]中收集了66 235 条氨基酸序列。在预训练结束后将BiLSTM 的权重冻结,在下游任务预测“药物-靶点”亲和力时就不会改变其权重。
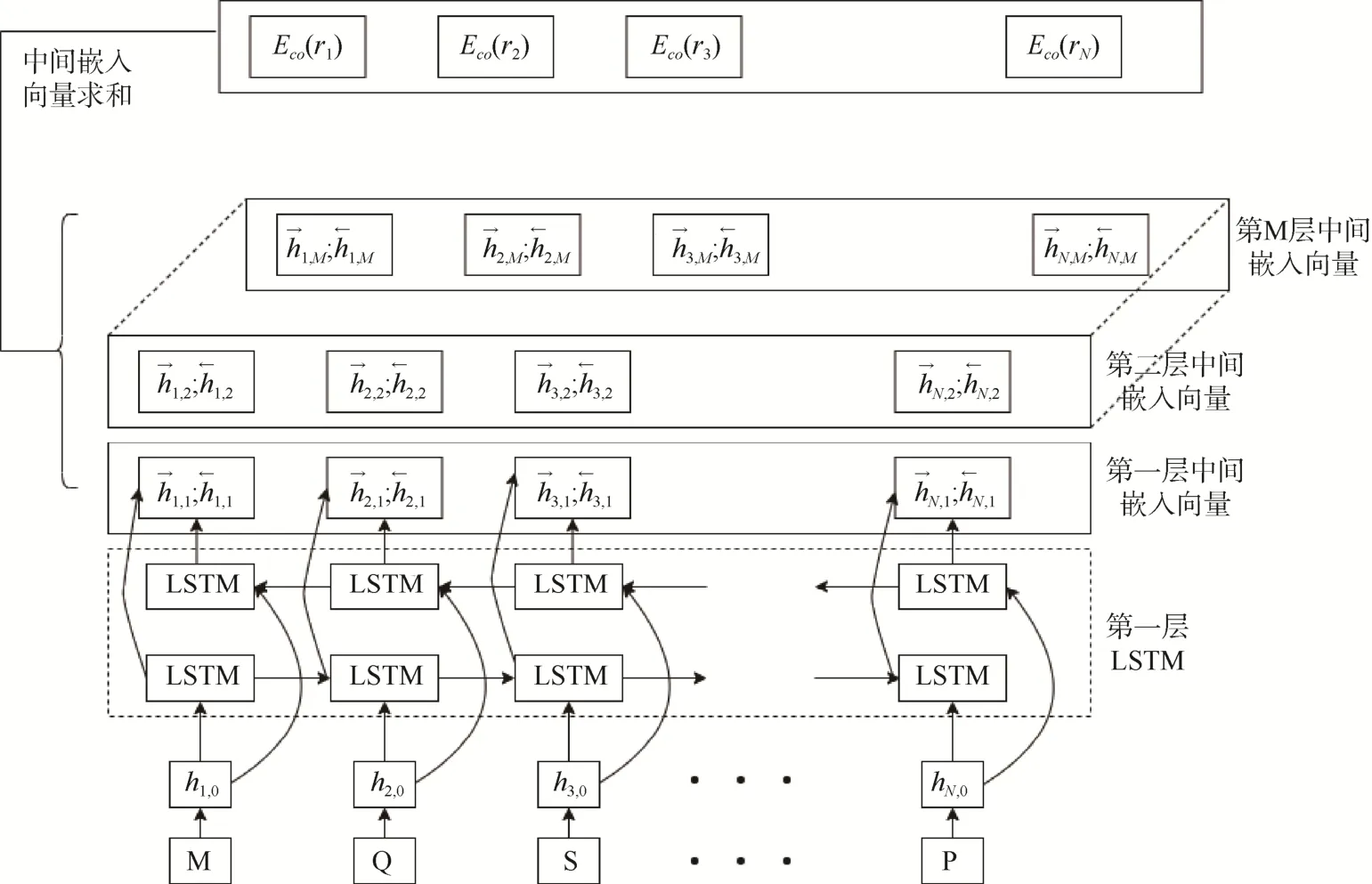
图2 BiLSTM 预训练模型网络结构
由于使用独热编码的SMILES序列和使用双向语言模型编码的氨基酸序列的长度不同,为了创建一个有效的表示形式,设定化合物的SMILES 的最大长度为100,氨基酸序列的最大长度为1 200,超过最大长度的化合物的SMILES序列和氨基酸序列将会被强制截断为最大长度。
1.3 预测网络模块
预测网络模块包含 2 个卷积神经网络(convolution neural network,CNN)模块及4 个全连接(fully connected,FC)层。2 个CNN 模块分别用于提取蛋白质和化合物的特征,4 个FC 层用于根据CNN 提取的特征预测蛋白质和化合物之间的亲和力。
1.3.1 CNN 模块
每个CNN 模块包含3 个堆叠的一维卷积层,每层卷积使用前一层的输出作为其输入。为了避免梯度消失问题,在每个卷积层上附加一个校正线性单元(rectified linear units,ReLU)。
1.3.2 FC 层
在每个CNN 模块后分别使用2 个全局最大池化层,分别为蛋白质和化合物生成高水平的特征向量,然后将2 个全局最大池化层的输出连接到4个FC 层。在FC 层中,前2 层各包含1 024 个神经元节点,第3 层包含512 个神经元节点,第4层仅包含1 个神经元节点。为了防止出现过拟合问题,在每个FC 层后面添加3 个速率为0.1 的随机失活(dropout)。计算蛋白质与化合物的相互作用分数,并通过Sigmoid 激活函数功能将相互作用分数调整成0~1 的数值。
为了训练给定的神经网络,使用平方根均误差目标函数作为损失函数,使用自适应矩估计算法优化网络参数[17],默认学习率为0.01。
1.4 数据集介绍
Davis 激酶结合亲和力数据集包含了激酶蛋白家族和相关抑制剂的选择性分析及其各自的解离常数值,里面含有442 种蛋白质及68 种化合物。KIBA 大规模激酶抑制剂生物活性数据集起源于一种叫做KIBA 的方法,它将不同来源的激酶抑制剂生物活性结合起来。KIBA 数据集最初有467 个目标和52 498 种药物,经过滤后,该数据集仅包含具有至少10 种相互作用的药物和靶点,总共产生229 种独特的蛋白质和2 111 种独特的药物。
1.5 实验环境
模型训练实验环境中的硬件设施主要为GeForce GTX2080Ti 型 GPU;软件设施主要为Ubuntu16.04 操作系统及Tensorflow 深度学习框架,其中Keras 的版本为2.2.5,算法实现语言采用Python 3.8。
1.6 模型评价
采用5 折交叉验证法分别在Davis 激酶结合亲和力数据集和KIBA大规模激酶抑制剂生物活性数据集上评估本文构建的“药物-靶点”亲和力预测方法的性能。将每个数据集中的“药物-蛋白质”对平均分成5 份,选择其中的4 份作为亲和力已知的“药物-蛋白质”对轮流训练本文提出的模型,将另外1 份作为亲和力未知的“药物-蛋白质”对用于预测亲和力,根据预测亲和力和真实亲和力计算均方误差和一致性指数,将其作为评价结果,平均5 次的评价结果为最终的评价结果,然后将该结果与使用KronRLS、SimBoost 和DeepDTA 算法的预测结果进行比较。
1.6.1 均方误差
使用均方误差(mean squared error,MSE)衡量预测的“药物-靶点”亲和力值和真实值之间的差距,计算公式如下。

式中,P表示预测值,Y表示真实值,N表示所有样本的个数。
1.6.2 一致性指数
使用一致性指数(concordance index,CI)衡量“药物-靶点”亲和力预测的性能[18],计算公式如下。
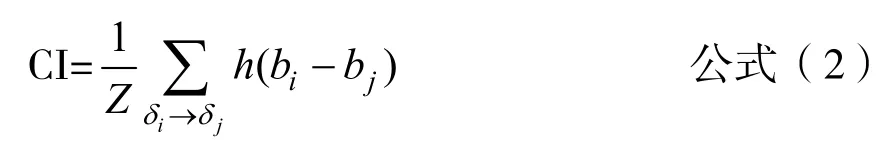
式中,bi表示大亲和力δi的预测值,bj表示小亲和力δj的预测值,Z表示一个归一化常数,h(x)表示阶跃函数。
2 结果
相较于KronRLS、SimBoost、DeepDTA,本文所构建的方法在Davis 激酶结合亲和力数据集和KIBA 大规模激酶抑制剂生物活性数据集上均获得了最高的CI 值和最低的MSE 值,见表1。
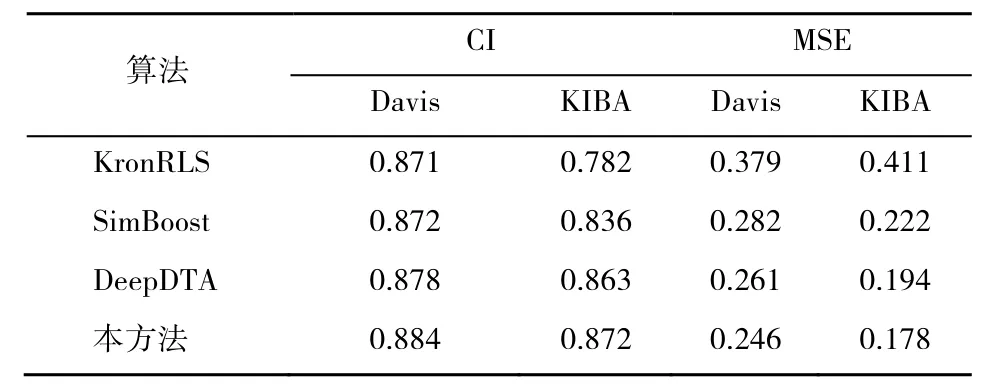
表1 4 种算法在2 个数据集上的实验结果
3 讨论
对于药物研发,找出候选药物与靶点之间的相互作用强度是至关重要的,而识别“药物-靶点”的相互作用已成为早期药物发现阶段的关键步骤。但是采用药物试验的方法进行药物筛选既昂贵又耗时。因此,迫切需要构建出能够以最小的错误率来识别潜在的“药物-靶点”相互作用的方法[19]。
根据已有研究[20]可知,药物和蛋白靶点呈三维结构,二者的结合是一个相对复杂的过程。本方法虽然不能完全反映出药物和靶点结合的复杂性,但三维结构不易获得,且已有相关文献论证了使用深度学习如DeepDTA 预测亲和力的有效性。本文在药物与靶点结合的三维结构不易获取的情况下,借助深度学习强大的非线性建模能力,仅使用蛋白质的氨基酸序列和药物的一维化学结构来预测“药物-靶点”亲和力,具有更强的适用性,其与静态氨基酸编码方式(如DeepDTA)的不同之处在于,预训练语言模型可以结合相邻氨基酸的信息动态对氨基酸序列进行编码,自动提取更为精细的氨基酸水平特征,让这些特征可以在不同的氨基酸序列上下游之间有所区别。此外,为了预测靶蛋白和药物之间的亲和力,本文设计了2 个独立的CNN 模块,从原始化合物序列和经过预训练语言模型编码的氨基酸序列中学习药物和蛋白质的特征,并将这些特征传送到一个全连接的网络中来预测亲和力。
本文比较了KronRLS、SimBoost、DeepDTA 算法及本方法在 Davis 激酶结合亲和力数据集和KIBA 大规模激酶抑制剂生物活性数据集上的MSE值和CI 值。其中MSE 值越低、CI 值越高,说明方法预测结果越准确。在Davis 激酶结合亲和力数据集中,SimBoost 和KronRLS 方法的性能类似;但是在KIBA 大规模激酶抑制剂生物活性数据集中,SimBoost 方法的CI 值高于KronRLS 方法的CI 值。KronRLS 是基于正则化最小二乘法的一种预测方法,其利用药物和靶点的相似矩阵来获得模型的参数值,在预测“药物-靶点”亲和力时仅依赖于药物和靶点的相似性,无法对复杂的药物和靶点的相互作用进行很好的预测。SimBoost 是一种基于特征工程的方法,需要专家来定义蛋白质和化合物的相关特征。DeepDTA 算法使用深度学习来挖掘蛋白质和药物的特征[21],因此其CI 值要高于KronRLS、SimBoos 这两种传统的方法。虽然DeepDTA 使用深度神经网络来模拟药物和靶点复杂的相互作用过程,但是其独热编码方法无法充分表达蛋白质的氨基酸序列信息。而本方法在2 个数据集上均获得了最高的CI 值和最低的MSE 值,说明本文使用的双向语言模型学习氨基酸序列信息较DeepDTA 仅使用独热编码的方式表达的信息更为准确,其预测能力要优于DeepDTA,且无须专业人员来定义蛋白质和化合物的相关特征,节约了人力资源及学习成本。
4 结语
预测药物与靶点之间的亲和力不仅可以提供更大的信息量,而且更具挑战性[22]。本文在药物与靶点结合的三维结构不易获取的情况下,借助深度学习强大的非线性建模能力,仅使用蛋白质的氨基酸序列和药物的一维化学结构来预测“药物-靶点”亲和力,且其预测结果的准确率高于KronRLS、SimBoost 和DeepDTA 方法。然而本文未考虑到药物的分子图结构信息,下一步研究将尝试将药物的分子图结构应用于“药物-靶点”亲和力的预测模型中,同时补充研究模型的可解释性,以期获得更满意的预测结果。
