2019年春季淮河中下游水体微生物的空间异质性
2022-11-17彭飞周彦锋王晨赫张希昭罗宇婷唐雪梅周依帆王东伟
彭飞,周彦锋,*,王晨赫,张希昭,罗宇婷,唐雪梅,周依帆,王东伟
(1.南京农业大学 无锡渔业学院,江苏 无锡 214081;2.中国水产科学研究院淡水渔业研究中心,农业部淡水渔业和种质资源利用重点实验室,江苏 无锡 214081)
淮河(31°55′~36°36′N,111°55′~120°25′E)是中国的七大河流之一,其下游主要流经的地域为安徽省和江苏省,为最典型的生态交错带流域之一[1]。流域中还包含中国第四大淡水湖——洪泽湖,该湖属平原水库型湖泊,西纳淮河,东入长江和黄海,对维持流域生态稳定作用显著[2]。近年来,淮河及其流域环境污染严重[3-7]。目前,有关淮河流域的研究多集中于水体水质的空间异质性和水生生物的种类及分布特征等方面,包括淮河流域夏季降水年代际变化特征及相应的大气环流异常[8]、淮河(安徽段)沉积物中微量元素的空间分布规律[9]、污染特征及潜在生态风险、近30年来淮河流域和洪泽湖水质的演变趋势[1]、淮河干流沉积物中重金属含量及分布特征[10]等。微生物是河流环境有机物分解的重要组成部分,对水体中的化学、物理和生态环境的变化非常敏感,其组成可以用于评估生态状况和水质。已有学者研究了长江中上游[11-12]、长江下游[13]和黄河[14-15]等中国主要河流流域中微生物的组成特征,而关于淮河微生物组成和分布特征的研究尚未见报道。淮河沿岸城市分布较为密集,其水质安全与人体健康、社会经济发展等密切相关,尤其是微生物污染风险越来越受到学者重视[16],对其微生物群落结构组成和分布差异及其环境影响因素的研究,有利于全面地评估淮河中下游的水质健康状况。
本研究中,以淮河中下游流域安徽-江苏段为研究对象,利用16S rRNA高通量测序技术分析水体中菌群的分布,探究不同生境菌群的空间差异及其环境影响因子,并评估水体污染的程度,以期为淮河流域内不同水环境的水体状况监测提供基础数据和参考依据。
1 材料与方法
1.1 研究区域及采样站位
研究区域位于安徽-江苏段淮河流域,于2019年3月5日—12日(枯水期)采集水样。采样点分4组:中游安徽段的3个断面,其附近的支流汇入繁杂,划为支流组(ZL),洪泽湖断面单独列为洪泽湖组(HZ),下游江苏段4个断面均位于主河道,划为河道组(YH),淮河入江口断面划为入江口组(HK)。9个采样点由上游至下游依次为阜阳南照(FY,32°36′32″N,116°00′31″E),凤台(FT,32°41′12″N,116°41′34″E),蚌埠闸(BB,32°57′34″N,119°09′10″E),洪泽湖(HZ, 33°17′07″N,117°17′05″E),高良涧闸前(GL,33°19′29″N,118°51′33″E),淮安闸前(HA, 33°28′14″N,119°09′31″E),淮安闸与邵伯闸之间(SB,33°08′33″N,119°22′51″E),邵伯闸与施桥闸(SQ,32°28′13″N,119°29′26″E),河道入江口(HK,32°16′53″N,119°28′39″E)(图1)。

图1 淮河流域中下游水体样本采集位点Fig.1 Collection sites of water samples in the middle and lower reaches of the Huaihe River Basin
1.2 方法
1.2.1 样品采集和水体理化指标测定 每个位点采集3个样品,全部27个样品均利用1 L无菌手持式柱状玻璃采水器采集,取0.5 L表层水环境样本,装入无菌玻璃瓶中,现场将样品过滤到孔径为0.22 μm的聚酯纤维滤膜上,将滤膜放入无菌的冻存管中置于液氮中保存,回到实验室后立刻转移至-20 ℃冰箱中保存,样品采集完成后两周内完成DNA样本的提取。

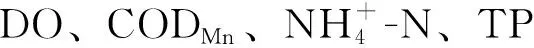
1.2.2 DNA的提取和高通量测序 将采集的滤膜剪碎,置于50 mL离心管中,按照DNA提取试剂盒说明书进行提取。采用E.Z.N.A.®Water DNA Kit(Omega Bio-tek,Norcross,GA,U.S.)提取基因组DNA,并用10 g/L琼脂糖凝胶电泳检测提取的DNA质量。选用细菌16S rRNA通用引物341F(5′ACTCCTACGGGAGGCAGCAG 3′)和806R(5′GGACTACHVGGGTWTCTAAT 3′)进行PCR扩增。PCR扩增程序:95 ℃下预变性2 min;95 ℃下变性30 s,55 ℃下退火30 s,72 ℃下延伸30 s共进行25个循环;最后在72 ℃下再延伸5 min。利用琼脂糖凝胶电泳检测PCR产物,判断扩增后的DNA浓度是否达到了试验扩增要求。在武汉华大医学检验所有限公司的Illumina Hiseq2500平台进行测序。
1.2.3 高通量测序结果初筛和数据分析 首先运用QIIME 1.8.0软件(quantitative insights into microbial ecology,http://qiime.org/)[19]识别疑问序列,除要求序列长度≥150 bp且不允许存在模糊碱基N之外,还将剔除5′端引物错配碱基数>1的序列和含有连续相同碱基数>8的序列。随后调用Usearch 5.2.236程序(http://www.drive5. com/usearch/)检查并剔除嵌合体序列。
所有数据均采用QIIME软件和R 3.2.0软件进行统计分析。通过Greengenes(Release 13.8)[20]数据库对所得到的OTU进行分类比对,采用QIIME软件获取各样本在门、纲、目、科和属5个分类水平上的组成和丰度分布,对OTU丰度矩阵中每个样本的序列总数在不同深度下随机抽样,以每个深度下抽取到的序列数及其对应的OTU数绘制稀释曲线,并对每个样本计算Alpha多样性指数,包括Chao1、Ace、Simpson和Shannon指数。通过Galaxy在线分析平台(http://huttenhower.sph. harvard.edu/galaxy/),对菌群相对丰度矩阵进行LEfSe分析,其中,LDA score=效应系数×标准化后的特征向量,并对该值进行对数转换。
1.2.4 富营养化评价 依据《湖泊(水库)富营养化评价方法及分级技术规定》(中国环境监测总站,总站生字[2001]090号),采用综合营养状态指数TLI(∑)评价各采样点的富营养化状况。综合营养状态分级标准:TLI(∑)>50时为富营养,50~60时为轻度富营养,60~70时为中度富营养,70~100时为重度富营养。
1.3 数据处理
试验数据均以平均值±标准差(mean±S.D.)表示,采用IBM SPSS Statistics 26.0软件对数据进行描述性统计分析和相关性分析,采用单因素(one-way ANOVA)和Tamhane’s T2法进行方差分析及多重比较(α=0.05)。采样图使用ArcGIS 10.2软件绘制,其他图表采用Origin 2017和OmicShare Tools软件绘制。
2 结果与分析
2.1 淮河中下游水质指标的空间差异性

水质评价结果显示,淮河中下游的水体污染源主要为氮污染。根据综合营养状态分级标准,可以看出,除入江口外的其余断面水质均有不同程度的富营养化,其中,阜阳达到了中度富营养(表1)。

表1 各站位环境因子平均值Tab.1 Average values of environmental factors in all sites
2.2 淮河中下游水环境菌群的空间差异性
2.2.1 菌群的多样性特征 经过质量检查和数据过滤后,从27个水环境样本中共获得945 846个序列,每个样品平均35 031个读数(每个样品 31 057~38 934个读数)。9个采样点的OTU数量和Alpha多样性指数见表2,各分组的特有OTU数分别为支流(31)、入江口(87)、河道(39)和洪泽湖(7),入江口的细菌群落丰富度高于其他3组,而多样性最高的是淮安闸,最低的是阜阳。
对淮河中下游微生物群落的多样性与环境因子进行Pearson相关性分析之前,对本研究中选取的15个环境数据进行共线性检验,其中,VIF>10时表示共线性显著,VIF为0~10时表示共线性不显著。在SPSS分析中剔除共线性显著的环境因子后,得到可以作为研究微生物多样性的影响因子。

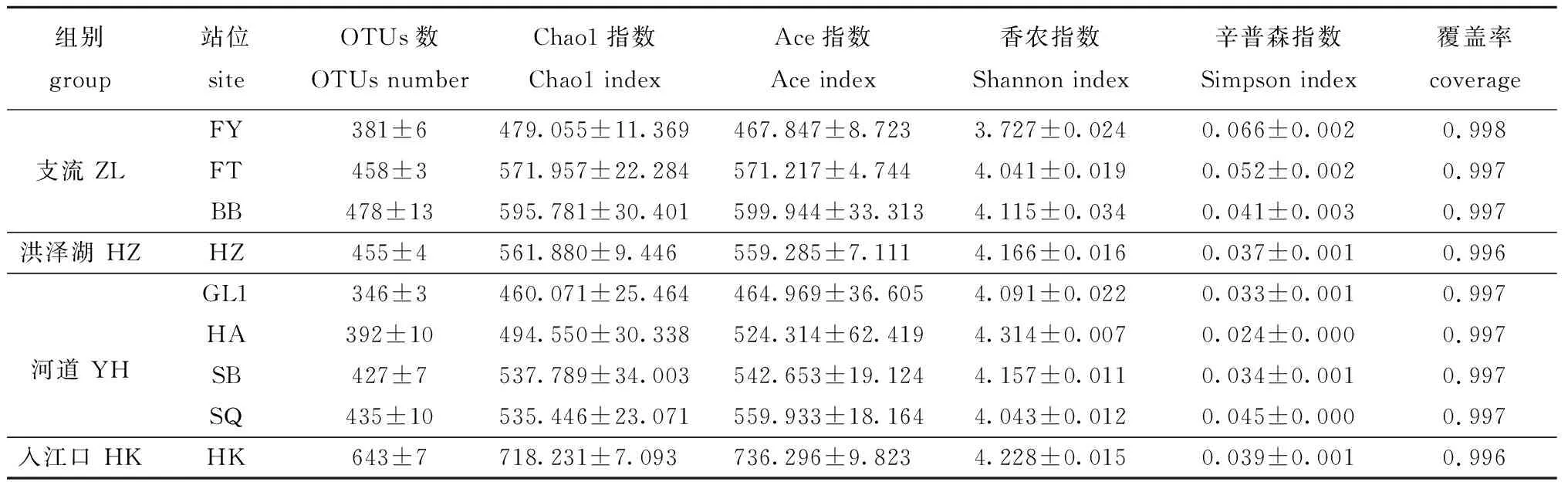
表2 水样细菌群落的丰度和Alpha多样性统计Tab.2 Statistics of abundance and alpha diversity of bacterial communities in water samples

表3 Alpha多样性指数与环境因子的相关关系(相关系数 R)Tab.3 Pearson correlation analysis between diversity index and environmental factors
2.2.2 微生物群落组成 将所有水体样本的OTU以97%的相似性阈值对其进行聚类,共鉴定出31门86纲148目229科326属细菌。各水体中微生物相对丰度排名前10的菌门如表4所示,其中,放线菌门Actinobacteria(各水体中相对丰度范围为30.1%~48.47%)、变形菌门Proteobacteria(25.36%~35.86%)和拟杆菌门Bacteroidetes(15.73%~31.76%)为所有水体样本中共同的优势类群,其余比例大于1%的门类细菌有2种,分别为疣微菌门Verrucomicrobia(2.05%~10.45%)和浮霉菌门Planctomycetes(0.59%~9.85%)。放线菌门主要由放线菌纲Actinobacteria(32.28%)组成,变形菌门主要由β-变形菌纲Betaproteobacteria(19.66%)、α-变形菌纲Alphaproteobacteria(4.90%)和γ-变形菌纲Gammaproteobacteria(3.53%)组成,拟杆菌门中黄杆菌纲Flavobacteriia和鞘脂杆菌纲Sphingobacteriia丰度最高。
每个分类单元在所有个体上的分布是不均匀的,门水平上,洪泽湖的浮霉菌门Planctomycetes丰度最高(P<0.05),变形菌门在蚌埠和入江口水样中相对丰度最高,分别为35.02%和32.68%,在其他站位的相对丰度范围为26.41%~29.76%,拟杆菌门在洪泽湖进入河道后丰度急剧增加,在淮安闸样点丰度最高(30.75%),至入江口时丰度又逐渐降低。入江口和支流样本中放线菌纲的丰度较高(35.63%, 37.10%),且入江口组的β-变形菌纲(25.75%)丰富度最高。浮霉菌纲Planctomycetia(8.5%)和Δ-变形菌纲Deltaproteobacteria(1.25%)在安徽段的相对丰度远高于下游江苏段,并且在洪泽湖中丰度最高,越往下游丰度越低。
从图2可见,在属水平上,黄杆菌属Flavobacterium、红育菌属Rhodoferax和Limnohabitans属在各位点中丰度最高,其余平均丰度超过1%的菌属有多核杆菌属Polynucleobacter、Fluviicola、沉积物杆状菌属Sediminibacterium和甲基娇养杆菌属Methylotenera等。河道组拥有发酵单胞菌属Zymomonas和甲基养菌属Methylibium两种特有的菌属,而HTCC、弓形杆菌属Arcobacter和突柄杆菌属Prosthecobacter则为河道组外的其他位点所特有。此外,阜阳的组线虫共生菌属Candidatus_Xiphinematobacter和Luteolibacter占比显著高于其他位点。图2中菌属对应的颜色越接近蓝色表示其丰度越低,颜色越接近红色表示其丰度越高。

表4 水体微生物在门水平上的相对丰度组成(相对丰度排名前10的菌门)
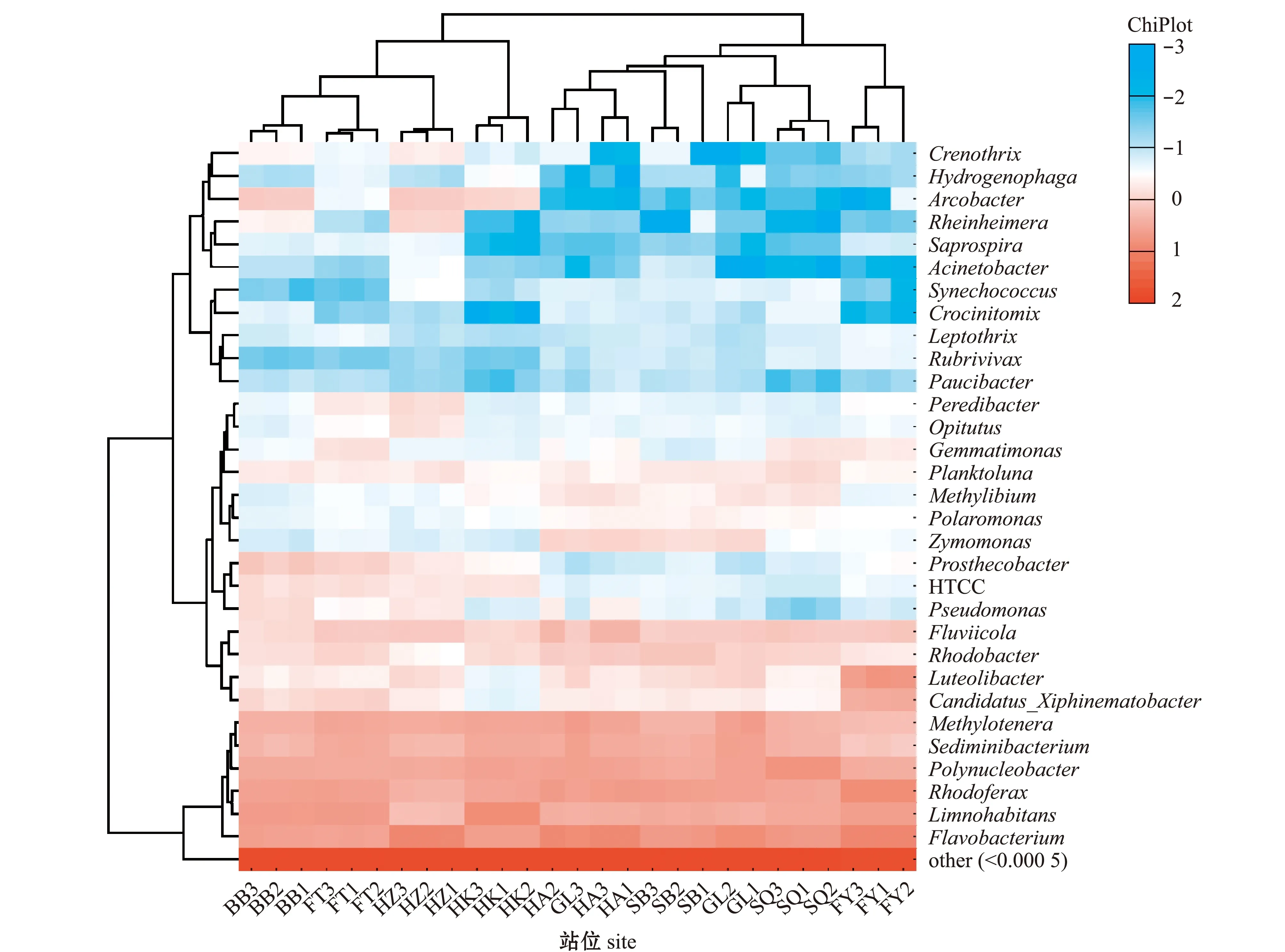
图2 各样本属水平菌群结构组成热图Fig.2 Heat map of the the structure and composition of bacterial flora at each sampling site at genus level
使用线性判别分析(LDA)效应量(LEfSe)算法比较了4组水体样本的微生物组成(相对丰度)的差异(图3)。根据多种假设检验校正的Wilcoxon非参数t检验,38个物种的OTU存在显著性差异(P<0.05),其中,支流和洪泽湖两组差异性菌群与其他两组相差较大。

图3 基于LEfSe分析的分类谱系图Fig.3 Taxonomic cladogram obtained from LEfSe analysis
2.3 环境因子与微生物群落的相关性分析
将各分组共有菌属中丰度排名较高和各位点特有的菌属与环境因子做Pearson相关性分析,结果如图4所示,其中绿色框中为环境因子与优势菌属的相关关系。

图4 属水平优势菌属与环境因子的Pearson相关性分析Fig.4 Pearson correlation analysis between dominant bacteria and environmental factors at genus level
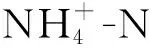
3 讨论
3.1 微生物群落的空间分布特征
自然水域中的微生物数量巨大,其对有机物降解及营养物质循环过程具有重要作用,是水生态系统的重要组成部分。全面了解水体微生物群落多样性、分布特征及其在生态系统中的功能和作用,对于管理和维护淮河中下游水生态环境具有深远的意义[21]。本研究中,采用 16S rRNA 高通量测序技术对淮河流域中下游(安徽-江苏段)水环境的细菌多样性进行探究,结果表明,放线菌门、变形菌门、拟杆菌门和浮霉菌门在各点位均有相对较高的丰度分布,这一结果也与其他关于淡水环境菌群的研究结果较为相似[22-23],这些类群与石油烃的降解和外来无机化合物的降解有较大关系[24],但不同空间分布的水体样品的细菌群落结构存在一定的差异性。厚壁菌门在淡水中也是较为常见的类群,但在本研究中其丰度极低,有研究表明,厚壁菌门与径流量呈正相关[25],2002年蚌埠闸扩建后淮河中下游河段流量的年平均极值降低,水流速度较为缓慢[1-2],推断可能是导致本研究中厚壁菌门丰度极低的主要原因。
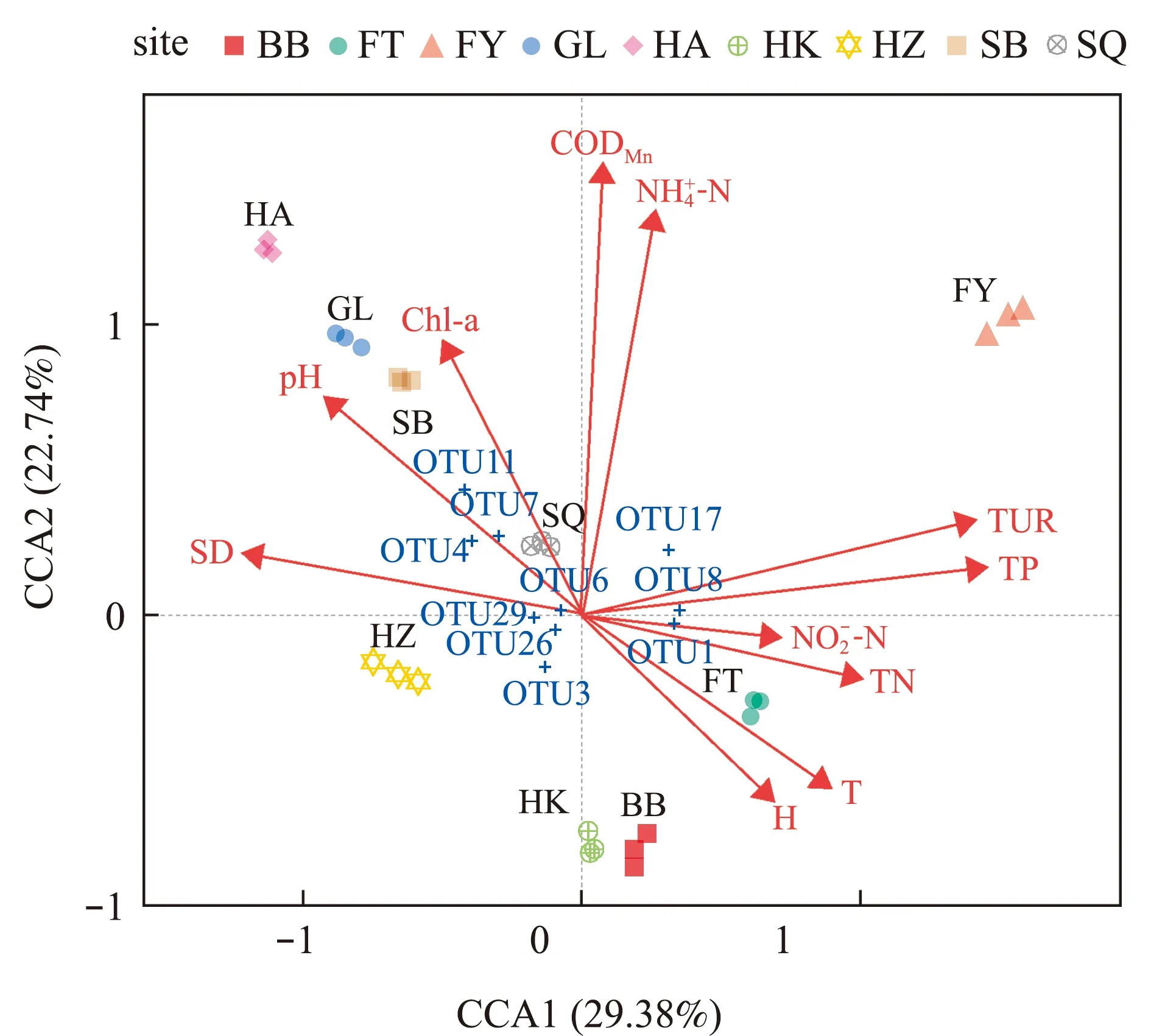
OTU1,OTU3 and OTU8—ACK-M1; OTU4—Gemmataceae; OTU6—Polynucleobacter; OTU7—Acidimicrobiales,C111;OTU11—Flavobac- terium;OTU17—Rhodoferax;OTU26,OTU29—Methylotenera_mobilis.图5 微生物群落结构与环境因子的CCA分析Fig.5 CCA analysis between microbial composition and environmental factors at the OTU level
安徽段支流中疣微菌门的相对丰度显著高于其他位点,此菌门一般在水环境或土壤中被发现,其中,疣微菌科Verrucomicrobiaceae多被发现在人类粪便中,同样气单胞菌科Aeromonadaceae也是人体和鱼类肠道中的病原菌,这可能与在淮河安徽段选取的3个位点沿岸受城市污水的影响较大有关。
洪泽湖中浮霉菌门的相对丰度较高,常发现该菌存在于污水处理系统中,另外,洪泽湖富含的黄杆菌属,常发现于酸沼泽及污水处理厂中,这与各种水体和土壤污染相关[26]。
河道中的高良涧闸、淮安闸和邵伯闸均位于淮安市,与其他位点不同,淮安闸的次级优势菌门为拟杆菌门,且由阜阳至淮安闸呈增长趋势,淮安闸位点丰度最高,蓝菌门的丰度趋势与拟杆菌门相同,这两种菌门均为淡水环境的常见优势菌门[27]。有研究表明,拟杆菌门丰度受蓝细菌影响较大[28-30],拟杆菌门中的类群可进一步沉降和分解有机物,还有研究表明,其中的黄杆菌科Flavobacteriaceae等异养细菌在聚合物的微生物降解中发挥着重要作用,这对于水生生态系统的健康发展具有非常重要的作用[28-30],它们还可以通过降解来源于蓝藻等藻类的有机质来获得自身生长的能量来源[31]。淮河中下游沿岸城市污水排放在一定程度上导致蓝藻等营养物质增多,从而影响拟杆菌门在水环境中的丰度。河道丰度较高的鞘脂单胞菌目通常在受污染的土壤中分离,其中一些物种能够利用多环芳烃(PAH)作为碳源,这种菌属的发现,可能与受到了船舶污染有关。发酵单胞菌属和甲基养菌属为河道特有的两种菌属,发酵单胞菌属具有MTBE(甲基叔丁基醚,Methyl tert-butyl ether)降解能力[32],甲基养菌属则是一种运用于乙醇发酵的菌属[33],这两种特有菌属的发现可能与河道沿岸的工业发展有关。
入江口中变形菌门和酸杆菌门丰度较高,变形菌门中的较多菌群是重金属污染土壤中的优势类群[34],酸杆菌门一般被认为是生活在重金属污染区域等酸性较强的环境中[35],这说明入江口可能存在酸化现象。另外,入江口还富集β-变形菌纲,它对环境具有较强的选择性,其中具有较多与反硝化相关的菌群,而伯克氏菌目Burkholderiales、从毛单胞菌科Comamonadaceae中的部分菌种均与重金属及石油污染相关[36-40]。由于入江口所处的特殊地理位置,以及多种因子间的交互作用,导致入江口处微生物多样性极其复杂,这种情况与渤海三湾有相似之处,均是受到了人类活动和河口输出的影响[41-42]。
3.2 微生物组成与环境因子间的关系
参照《地表水环境质量标准》可发现,本次采样各位点中的氮含量相对较高,同时结合微生物组成的结果,黄杆菌属为反硝化细菌[43],红育菌属为异养脱氮菌[44],Limnohabitans属的作用是参与氮循环和分解蓝藻[11],这3种菌属为各分组丰度排名前3的共有菌属,其他占比超过1%的菌属Fluviicola(利用硝态氮)、沉积物杆状菌属[45]和甲基娇养杆菌属(脱氮菌属),它们的功能均指向水体中的氮利用。微生物群落也是水体富营养程度的重要生态指标,放线菌门在各采位点中均占主导地位,这与本次调查中普遍存在的水体富营养化密切相关[46]。

4 结论
1)淮河中下游水体普遍存在富营养化情况,水质指标评价中总氮浓度普遍偏高,以及多种丰度较高的菌属与氮的相关关系,反映了各采样点的氮污染情况。
2)放线菌门、变形菌门和拟杆菌门是所有位点中共同的优势菌门,而安徽段疣微菌门相对丰度较高,洪泽湖中浮霉菌门的相对丰度较高,入江口中变形菌门和酸杆菌门相对丰度较高;黄杆菌属、红育菌属和Limnohabitans属在各位点中丰度最高,发酵单胞菌属和甲基养菌属是河道组特有的菌属,而HTCC、弓形杆菌属和突柄杆菌属则是入江口、支流和洪泽湖3个生境组所特有。优势类群的差异与不同生境组的污染状况及地理位置密切相关。
