一个常染色体隐性遗传性耳聋家系中MYO7A基因的突变与遗传分析
2022-11-10李萌孙金仓健刘敏文卫平
李萌,孙金仓健,刘敏,文卫平,2
(1.中山大学附属第一医院 耳鼻咽喉科 中山大学耳鼻咽喉科研究所,广东 广州 510080;2.中山大学附属第六医院 耳鼻咽喉科,广东 广州 510655)
耳聋是最常见的感觉功能障碍性疾病之一,新生儿中的发病率约1‰~3‰。耳聋病因复杂,约60%与遗传因素相关[1]。遗传性耳聋是一种高度异质性的疾病,根据是否伴有其他系统疾病可分为综合征型耳聋(30%)和非综合征型耳聋(70%)。非综合征型耳聋更为常见,其中75%~80%表现为常染色体隐性遗传,多表现为先天性极重度感音神经性聋[2]。MYO7A基因位于染色体11q13.5,编码由2 215个氨基酸构成的非常规肌球蛋白ⅦA,广泛表达在视网膜上皮和内耳毛细胞静纤毛束中[3]。MYO7A基因变异所致耳聋表型具有高度的异质性,既可导致超过50%的1型Usher综合征型耳聋[4],也可导致常染色体显性遗传性耳聋11型(deafness,autosomal dominant 11, DFNA11)[5]和常染色体隐性遗传性耳聋2型(deafness,autosomal recessive 2, DFNB2)[6]。
MYO7A基因自1996年报道至今,已被证实有500多个突变位点与遗传性耳聋相关[7]。本研究通过高通量测序对一个先天性耳聋家系进行基因型与表型分析,发现该家系的可能致聋突变为MYO7A未曾报道的复合杂合突变NM_000260.3:c.765C>A(p.F255L)和c.275_278dupACCT(p.I94fs),两个突变位点均为首次报道的MYO7A新的变异位点,丰富了MYO7A基因的变异谱,现总结报道如下。
1 资料与方法
1.1 研究对象
研究对象为一个常染色体隐性遗传的核心家系,包括2代4人,3名正常人,1例耳聋患者(家系先证者Ⅱ-2)。先证者为1岁11月女童,因发现对声音反应差1年余,就诊于中山大学附属第一医院耳鼻咽喉科门诊,经详细听力学检查确诊为双耳极重度感音神经性聋,并行耳聋基因检测。
1.2 研究方法
1.2.1 临床资料采集 本次研究获得中山大学附属第一医院医学研究伦理委员会的批准,家系成员均签署知情同意书,未成年成员由家长代为签署。由我科2名副主任以上医师对家系成员进行了详尽的病史采集和全身体格检查,并填写了耳聋信息调查表,排除药物、噪声及其它因素造成的耳聋,对先证者父母及姐姐(6岁女童)进行了纯音听阈和声导抗测试,对先证者进行了声导抗、畸变产物耳声发射(distortion product otoacoustic emission,DPOAE),听性脑干反应(auditory brainstem response,ABR),多频稳态(auditory steady-state response,ASSR)及颞骨高分辨CT以及中内耳MRI检查。采集家系成员外周血5 mL用以提取外周血DNA并进行高通量测序。
1.2.2 高通量测序及生物信息学分析 高通量测序由广州金域医学检验中心完成,对遗传性耳聋518个相关基因的全部外显子及剪接区进行靶向捕获测序。将家系先证者Ⅱ-2基因组DNA片段化(180-280bp),制备DNA文库,通过基因芯片对目标基因外显子及邻近剪接区DNA进行捕获和富集,使用Illumina HiSeq平台进行高通量、高深度测序(平均测序深度200×)。
对原始测序数据进行测序数据的质量评估,将符合标准的测序数据比对到参考基因组(GRCh37/hg19)上,检测样本的变异信息,并对变异数据进行统计和注释。通过基因组分析工具包(genome analysis toolkit,GATK)来检测分析单核苷酸变异(single-nucleotide variants,SNV)和InDel变异,使用CNVnator v0.2.2软件分析发现拷贝数变异(copy number variants,CNVs),利用ANNOVAR软件对变异信息进行注释,包含dbSNP数据库、1 000 g数据库、国家心肺和血液研究所外显子组测序计划(NHLBI-ESP project,esp6500si_all数据库)、外显子组聚集联盟(Exome aggregation consortium,ExAC)和其他已有的数据库的注释信息(clinvar,HGMD等),注释内容涵盖变异的位置信息,类型,保守性预测等。过滤已报道的单核苷酸多肽性(single nucleotide polymorphism,SNP),同义变异(文献报道的致病变异除外),以及正常人数据库中频率>0.01的变异(GJB2和SLC26A4除外)。
1.2.3 Sanger测序验证 使用UCSC在线数据库(https://genome.ucsc.edu/) 查询变异位点附近的参考序列并以此为DNA模板,使用Primer3在线引物设计软件(http://bioinfo.ut.ee/primer3-0.4.0/) 设计PCR引物,扩增包含变异位点的DNA片段,并对扩增产物进行琼脂糖凝胶电泳,检测PCR产物的丰度和特异性,将符合测序标准的PCR产物进行Sanger测序,验证变异位点,并在家系内进行基因型与表型分析。
2 结果
2.1 家系遗传特征及临床表型分析
该家系为一个两代常染色体隐性遗传核心小家系(图1),未诉家族耳聋病史,包含1例耳聋患儿(Ⅱ-2),1名具有正常听力表型的子代(Ⅱ-1)和双亲(Ⅰ-1、Ⅰ-2)。Ⅱ-2出生时听力筛查结果不详,家长未诉眼科相关异常,体格检查及生长发育未见明显异常,无特殊用药史、外伤及噪音暴露史,视力、视野及眼底检查不能配合。声导抗示双耳A型曲线,DPOAE双耳各频率未引出,ABR及ASSR示双耳极重度感音神经性聋。乳突HRCT及中内耳MRI未见明显异常。Ⅰ-1、Ⅰ-2、Ⅱ-1体格检查及眼科检查(视力、视野、眼底)均未见明显异常,声导抗均为A型曲线,纯音听阈测试听力正常。
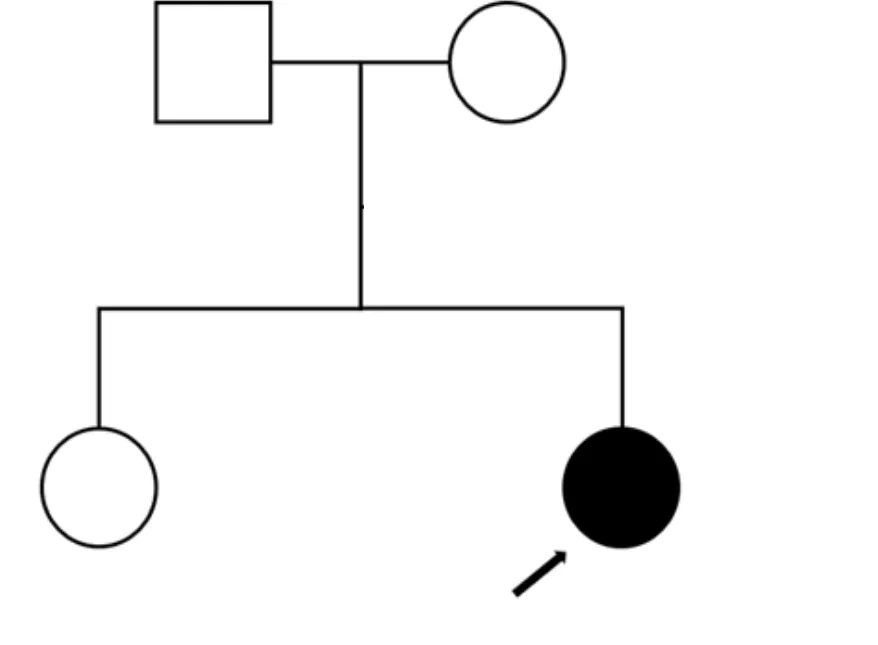
图1 家系图 黑色箭头标注成员为家系先证者;*标注成员进行高通量测序
2.2 基因变异检测
高通量测序发现先证者MYO7A基因存在NM_000260.3:c.765C>A(p.F255L)和c.275_278dupACCT(p.I94fs)2个候选致病变异。经Sanger测序验证示c.765C>A突变遗传自父亲,c.275_278dupACCT遗传自母亲,先证者姐姐为c.765C>A杂合携带,在家系中耳聋表型与MYO7A基因型共分离(图1、2)。
2.3 MYO7A基因变异致病性分析
c.275_278dupACCT为移码突变,在HGMD、esp6500si_all、1 000g数据库、gnomAD_all、ExAC_all、dbSNP数据库中均未见收录(PM2),导致翻译蛋白氨基酸序列自94位开始移码并提前终止(图3、4),该变异预计会导致所编码的蛋白发生截短而失去正常功能(PM4),依据ACMG变异分类指南[8-9],该变异为致病性变异(PM2+PM4)。
c.765C>A为错义突变,仅在dbSNP数据库中收录(rs782702948),为超稀有变异(PM2);多个变异有害性预测软件[SIFT、PolyPhen2、Provean、LRT and CADD (the combined annotation dependent depletion)]预测均为有害或致病性(PP3)(图4);该变异位于明确缺少良性变异的功能结构域中,位于变异热点区域(PM1)(图3);经分析该家系成员的基因检测结果与临床表型符合基因型-表型共分离PP1-S(PS),根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)变异分类指南综合分析,c.765C>A可判为可能致病性变异[PM1+PM2+ PP1-S(PS)+PP3]。
3 讨论
MYO7A具有高度的遗传异质性和基因多效性。MYO7A基因突变既可导致非综合征型耳聋(DFNA11和DFNB2),也可引起同时伴有内耳和视网膜病变的综合征型耳聋(Usher综合征1型)。DFNA11的主要临床表现为迟发性、渐进性语后聋;DFNB2常表现为重度或极重度的语前或语后聋;Usher综合征1型为常染色体隐性遗传,主要临床表现为先天性重度或极重度感音神经性聋,早发性视网膜色素变性,还常合并前庭功能异常[7]。本次研究的核心家系,双亲和姐姐表型正常,无听力和视觉障碍,无头晕、耳鸣主诉,先证者为1岁11个月女童,双耳极重度感音神经性聋,言语发育差,视力、视野及眼底等眼科检查因患儿年幼不能配合而尚不明确,该家系为常染色体隐性遗传方式,尚不能排除Usher综合征型耳聋。
MYO7A基因位于染色体11q13.5(图3),于1995年由Wei等首次报道,包含49个外显子,编码2 215个氨基酸组成的蛋白MyosinⅦA,属于肌球蛋白家族成员,MyosinⅦA为马达蛋白,具有ATP酶活性,能够利用水解ATP产生的能量与肌动蛋白结合,并沿着肌动蛋白丝运动[10]。MyosinⅦA蛋白结构包含3个重要区域(图3):①氨基末端的马达区域,主要功能是水解ATP并产生动能,与肌动蛋白结合,是肌球蛋白的核心功能区域;②颈部的调节区,与肌球蛋白轻链结合形成异亮氨酸-谷氨酰胺基序;③尾端的羧基末端,主要作用为调节MYO7A运动,包括SH3结构域、2个FERM结构域和2个MyTH4结构域[11]。MyosinⅦA蛋白在内耳和视网膜均表达,内耳中主要分布在毛细胞静纤毛、细胞体和表皮板,对维持毛细胞机械电转换通道的紧张度必不可少,还影响毛细胞静纤毛的形态和功能[12],因此,蛋白功能的改变将导致不同程度的听觉及前庭功能障碍[13]。目前已报道500余个MYO7A致聋
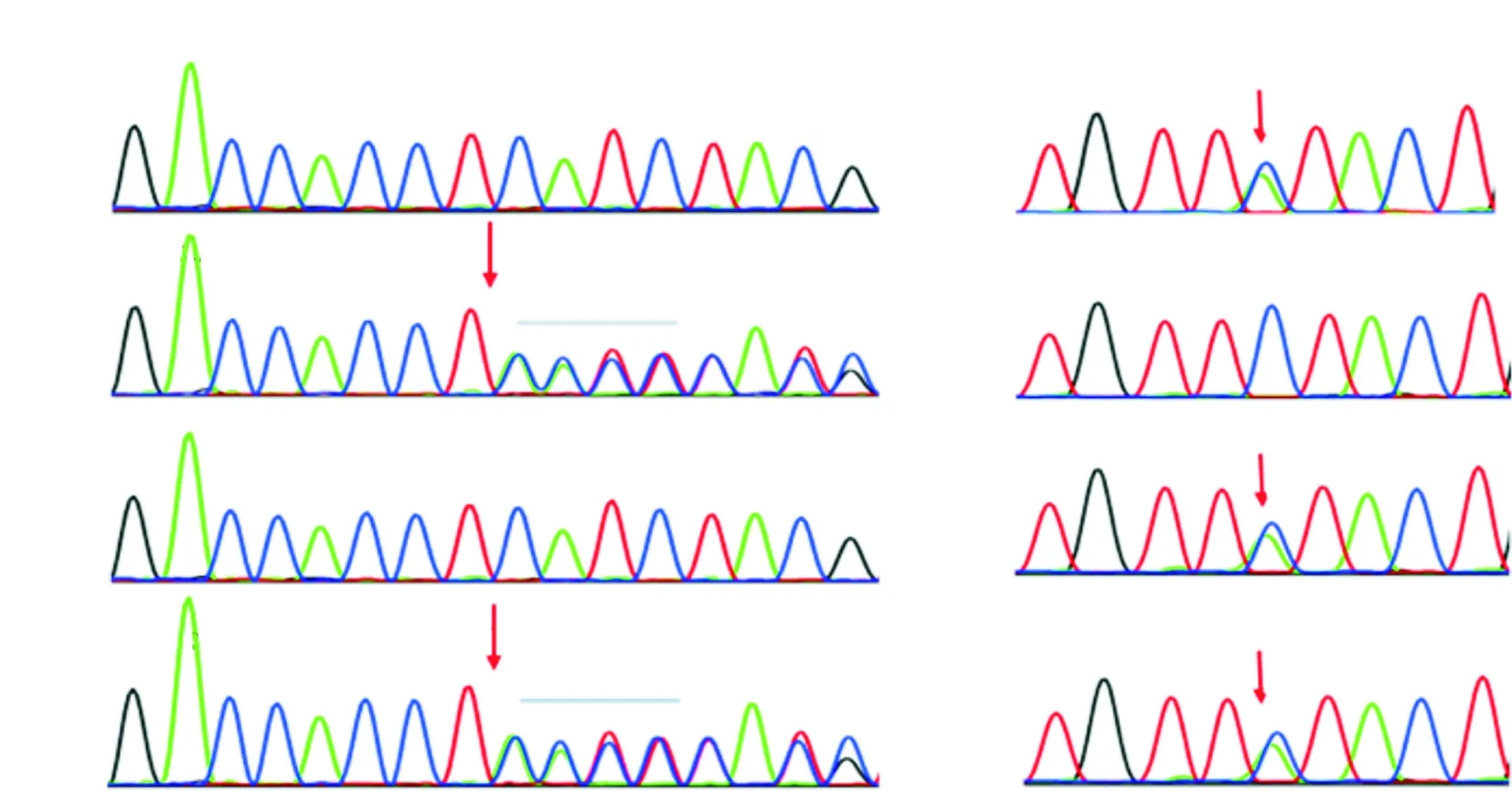
图2 MYO7A c.275_278dupACCT (a)和c.765C>A (b)在家系成员中的Sanger测序图
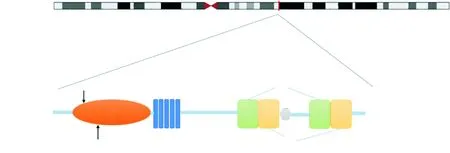
图3 MYO7A基因结构及p.I94和p.F255两个突变在MyosinⅦA蛋白结构示意图中的位置

图4 多个物种间MYO7A基因突变位点氨基酸序列保守性分析
本研究通过对先证者行高通量测序,靶向518个耳聋相关基因的全部外显子及剪接区发现先证者的MYO7A基因存在NM_000260.3:c.765C>A(p.F255L)和c.275_278dupACCT(p.I94fs)两个突变位点。通过Sanger测序在家系中进行基因型分析,发现先证者为c.765C>A和c.275_278dupACCT复合杂合突变,突变分别来自父亲和母亲,姐姐携带c.765C>A突变,变异在家系中与表型共分离(图1、2)。c.275_278dupACCT为移码突变,未在人群及相关临床病例中报道过,导致编码氨基酸序列自94位开始移码并提前终止,预计会使MyosinⅦA蛋白出现截短而失去正常功能,依据ACMG变异分类指南,该变异为致病性变异(PM2+PM4)。c.765C>A为错义突变,也未在人群及相关临床病例中报道过,变异位于蛋白的马达区域(图3),序列在多个物种中高度保守(图4),为该基因的变异热点区域,多个变异有害性预测软件预测变异会影响蛋白功能,并在家系中与表型共分离,根据ACMG变异分类指南综合分析,c.765C>A可判为可能致病性变异(PM1+PM2+ PP1-S(PS)+PP3)。综合以上高通量测序、变异致病性分析以及基因型-表型验证,c.765C>A和c.275_278dupACCT可能是MYO7A基因的新致病位点,目前在国内外文献中尚未报道。本次研究丰富了我国遗传性耳聋的基因变异谱,为遗传性耳聋的诊断、产前咨询提供了新的参考。
该患儿为极重度感音神经性聋,言语发育差,乳突HRCT及中内耳MRI未见明显异常,在我院已行右侧人工耳蜗植入术,术中监测示右耳神经反应遥测技术反应好,术后患儿正常开机,并按计划进行听力及言语康复中。因此,建议对此型基因变异患儿尽早行人工听觉干预,以促进听力及言语康复。现患儿年幼无法配合眼底及视野检查,眼部情况尚不明确,不能排除Usher综合征,建议对该患儿的视力、视野及眼底情况进行动态监测,并及时干预保护患儿视力。遗传性耳聋致病基因检测和诊断技术的发展,不仅能够从基因型的角度预测临床表型,指导临床诊治;而且能够对遗传性耳聋家庭提供遗传咨询、产前诊断及出生干预,有效避免再次生育耳聋患儿,对遗传性耳聋的三级预防具有重要指导意义。
