聚合物相变储能微胶囊的制备研究
2022-11-09王建斌王若磊
王建斌, 王若磊
(1 宁夏银帝地产集团有限公司,宁夏 银川 750021;2 宁夏现代建设监理有限公司,宁夏 银川 750000)
相变储能材料的能量变化通常是通过固相和液相之间的相互转化实现的[1-2]。当外界环境变热时,PCM由固态熔化为液态,需要吸热;反之,当外界环境冷却下来,PCM由液体凝结成固体,需要放热。相变材料因其熔融和凝固温度范围宽、储能密度高而被用作重要的储能形式[3-5]。
微胶囊技术是由物理、化学或物理化学的方法,通过使用无机或者聚合物材料包裹固、液、气功能材料的壳核构造的固体粒状物[6-8]。这些固体粒状物的粒径一般位于5 nm~1000 μm。目前,微胶囊技术发展迅猛,据统计已有200多种制备方法[9-12]。近年来,又出现了许多新颖的制备方法,如逐层自组装法、乳液聚合法、声化学法等[13-15]。
本文以正十八烷(相变材料)为芯材,以聚甲基丙烯酸甲酯为壁材,通过声化学方法和两步聚合技术——乳液聚合和原位聚合法制备了相变储能微胶囊。通过DLS、SEM、TGA、DSC、XRD等手段对制备的聚合物相变储能微胶囊的物相、形貌、热性能等进行了系统测试表征。
1 实 验
1.1 仪器与试剂
HWCL-1型磁力搅拌器,郑州长城科工贸有限公司;PCD-2000型恒温鼓风干燥箱,上海琅玕实验仪器公司;DX-2700型X射线衍射仪,上海精密科学仪器有限公司;VCX-750型超声波细胞粉碎仪,美国SONICS&MATERIALS公司;扫描SIGMA-500型扫描电子显微镜,德国Zeiss公司;TA-Q50型热失重分析仪,美国TA公司;TA-Q20型差示扫描量热仪,美国TA公司;Nano-ZS90型激光粒度及电位分析仪,英国Malvern仪器有限公司。
甲基丙烯酸甲酯(MMA),甲基丙烯酸烯丙酯(AMA),正十八烷(n-Oct),偶氮二异丁腈(AIBN),苯乙烯-马来酸酐共聚物钠盐(SMA),所有化学试剂均为分析纯级,所有用水均为二次蒸馏水。
1.2 实验方法
1.2.1 微胶囊的制备
称取10.0 g SMA和200.0 g去离子水倒入500 mL烧杯中,用玻璃棒搅拌使其均匀溶解,然后放入50 ℃水浴锅中,恒温10 min。准确称取10.0 g MMA、3.0 gAMA、12.0 gn-Oct倒入100 mL烧杯中,加入1.0 g AIBN,搅拌使其溶解,配成油相,倒入水相混合。
将上述混合液分成两份,一份未乳化直接置于超声装置中反应;另一份置于高速剪切乳化机上,以8000 r/min速率剪切10 min得到油/水(O/W)乳液,再置于超声装置中反应。
1.2.2 超声聚合反应
将油水混合物倒入超声反应瓶,然后将超声探头插入液体至油水边界处,采用脉冲方式(超声2 s、停顿2 s),超声时间为30 min,抽滤得到微胶囊固体粉末,冻干待用。
1.2.3 测试
(1)扫描电镜:采用蔡司SIGMA-500型扫描电镜对微胶囊形貌进行观察。同上,取稀释后的微胶囊悬浮液滴至硅片上,室温干燥后观察。
(2)热失重:采用TA-Q50型热失重分析仪对微胶囊进行热失重测试。温度区间:室温至600 ℃,升温速率10 ℃/min。
(3)差示扫描量热:采用TA-Q20型差示扫描量热仪对微胶囊进行测试。温度区间:-10~50 ℃,升温速率5 ℃/min。
(4)X射线衍射:采用DX-2700型X射线衍射仪对微胶囊进行测试。铜靶:λ=0.15418 nm(Kα射线),扫描范围10°~80°,扫描速率2°/min。
2 结果与讨论
2.1 反应条件对微胶囊粒径的影响
选取反应条件不同的三组微胶囊悬浮液进行粒径分析发现,直接进行幅值为50%超声得到的微胶囊粒径大小为540.2 nm,乳化后再进行幅值为50%超声得到的微胶囊粒径大小为650.8 nm。一般情况下,聚甲基丙烯酸甲酯为外壳的微胶囊粒径分布在0.5~1.5 μm之间,间接证明了聚甲基丙烯酸甲酯微胶囊外壳已经形成。由粒径可以看出,随着反应条件的不同,直接超声聚合得到的微胶囊粒径反而更小,且乳化反应会消耗乳化剂并增加了反应步骤,因而声化学反应优势明显。
2.2 反应条件对微胶囊形貌的影响

图1 不同制备条件下微胶囊的SEM照片Fig.1 SEM images of microcapsules under different conditions
如图1所示,聚甲基丙烯酸甲酯微胶囊均呈现出比较规则的球形,且表面光滑。图1a中,直接进行超声反应得到的微胶囊粒径大小约为100 nm,此直径小于动态光散射测得的粒径,原因在于,DLS测得的是悬浮液中微胶囊的水力学粒径,而SEM测得的是经烘干后的微胶囊粒径,二者自然有较大差异。图1b中,乳化后再进行超声反应得到的微胶囊与图1a相似,说明直接进行超声反应即可简便、快速地制备聚合物相变储能微胶囊。
2.3 微胶囊的DSC曲线分析
图2为正十八烷微胶囊的DSC曲线。纯十八烷在26~34 ℃发生熔融相变,熔融温度为30.3 ℃,熔融相变热为334.6 J/g。由图2可知,正十八烷微胶囊在27~35 ℃发生熔融相变,熔融温度为31.1 ℃,熔融相变为178.9 J/g,结晶相变为181.1 J/g。通过比较发现,微胶囊的熔融焓值和结晶焓值低于纯十八烷,这是因为在加热过程中,微胶囊芯材发生相变,而不发生相变的聚甲基丙烯酸甲酯壁材的加入对焓值没有贡献,使得总焓值降低。熔融温度方面,聚合物微胶囊与纯十八烷相比变化不大,即壁材对此几无影响。
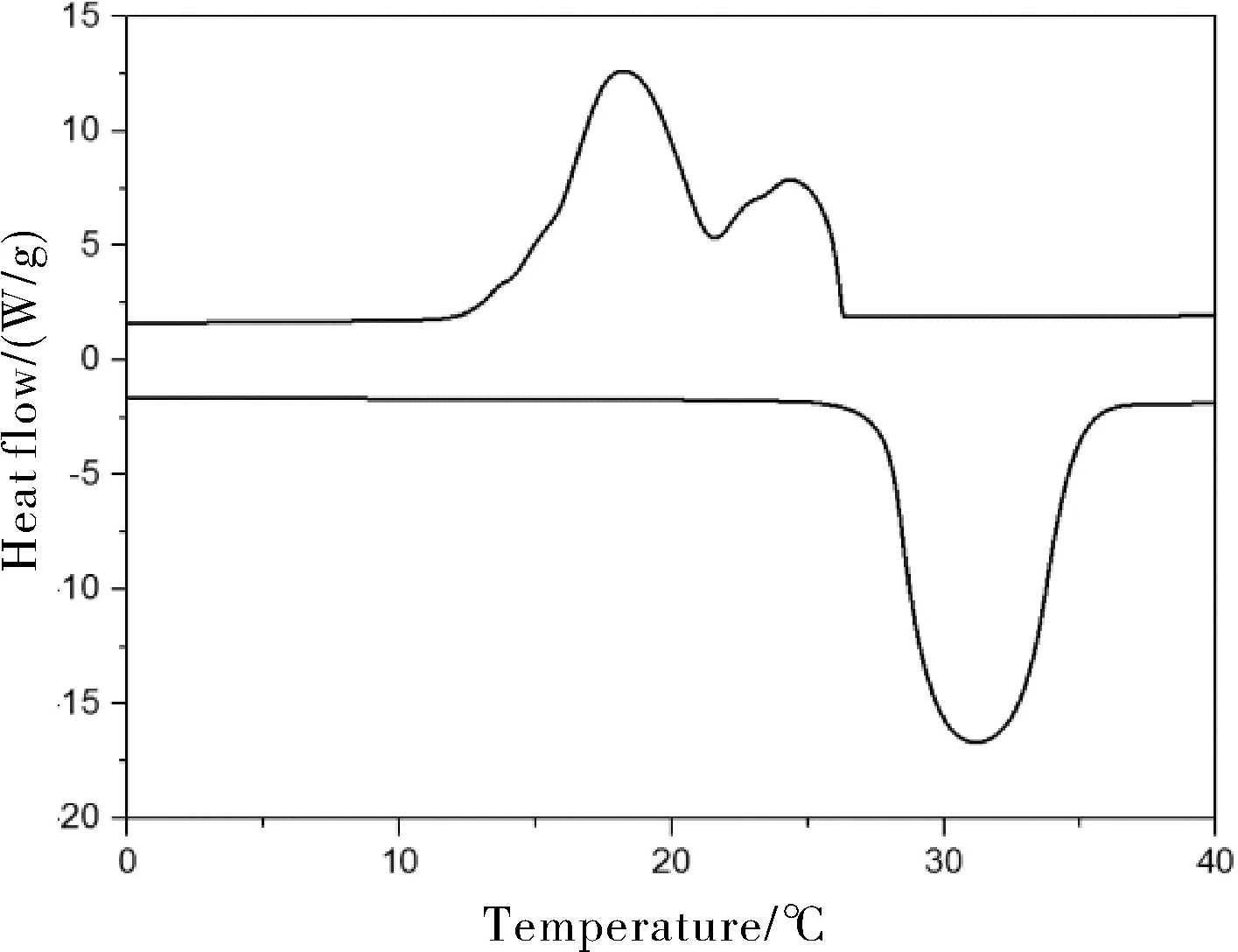
图2 微胶囊的DSC曲线Fig.2 DSC curves of microcapsules
2.4 微胶囊的TG曲线分析
图3为正十八烷微胶囊的TG曲线。正十八烷在127~219 ℃范围内有明显的失重现象,直至235 ℃已基本分解。而在正十八烷微胶囊的失重曲线中,除去初始阶段水分的挥发,正十八烷微胶囊的失重过程大致分为三个阶段:第一阶段发生在130 ℃左右,这是由于正十八烷发生了氧化分解,失重率为65%;第二和第三阶段发生在200~400 ℃之间,这是由于壁材发生分解所致,失重率接近90%。相对于纯的正十八烷来说,热失重现象发生了延迟,说明壁材延缓了正十八烷的分解,提高了正十八烷微胶囊的热稳定性。
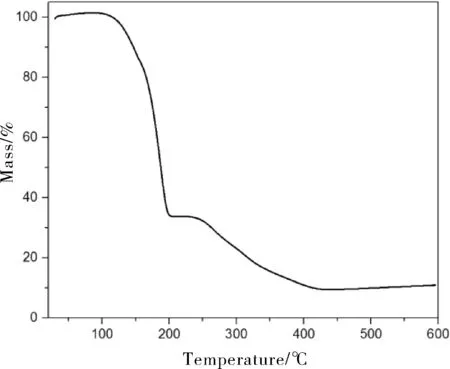
图3 微胶囊的TG曲线Fig.3 TG curve of microcapsules
2.5 微胶囊的XRD曲线分析
图4为正十八烷微胶囊的XRD曲线,图5为纯正十八烷的XRD曲线。正十八烷的特征峰出现在2θ=19.42°、22.34°、23.52°、24.76°、34.66°、38.92°和44.74°处,这些特征峰也存在于正十八烷微胶囊中。需要说明的是,相比正十八烷来说,这些特征峰的位置有些小的移动,特征峰的强度也有所降低。这是由于,正十八烷微胶囊化以后,分子运动受限,结晶度降低,这也间接证明了聚甲基丙烯酸甲酯实现了对正十八烷的包覆。
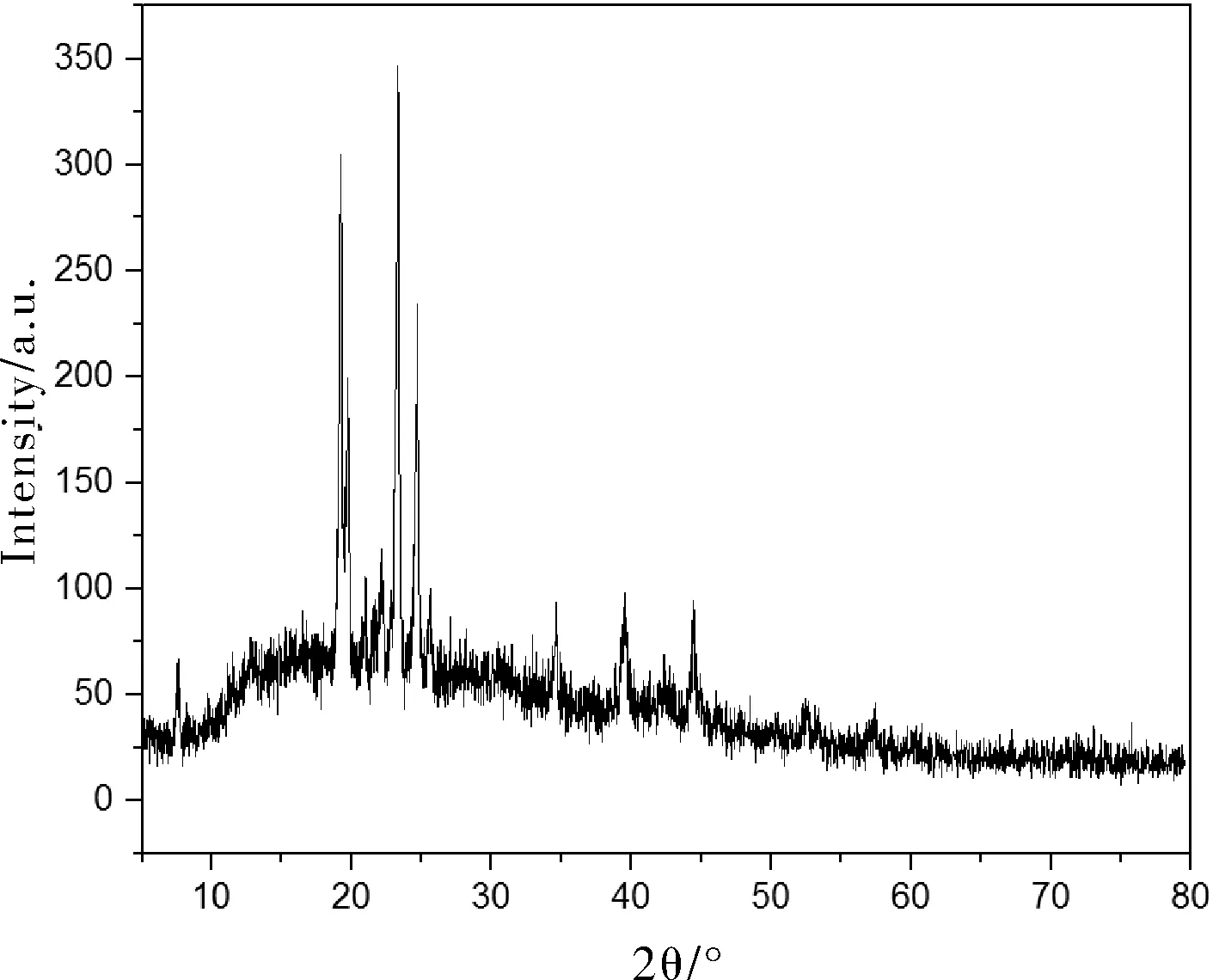
图4 微胶囊的XRD谱图Fig.4 XRD spectrum of microcapsules
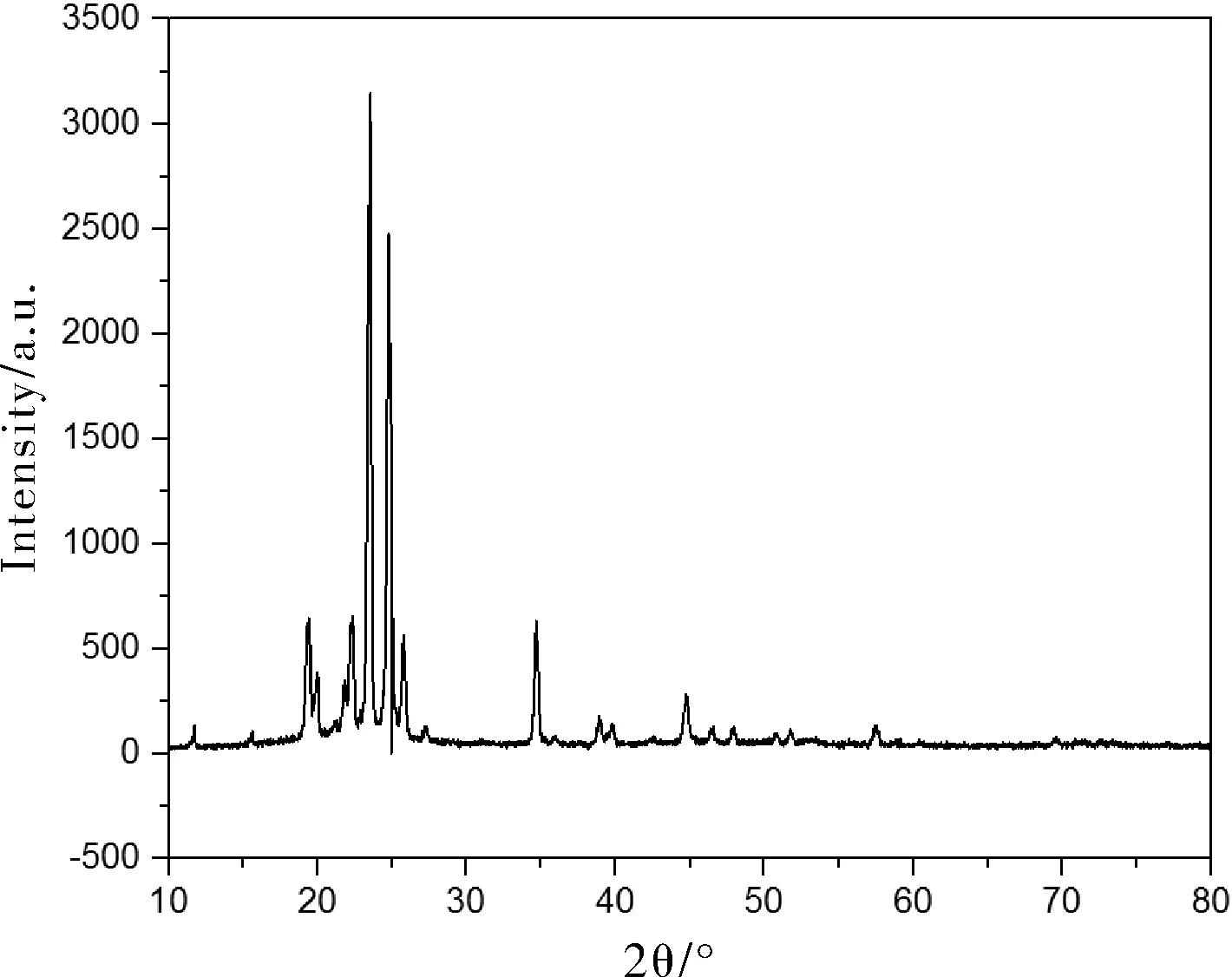
图5 正十八烷的XRD谱图Fig.5 XRD spectrum of octadecane
3 结 论
本文采用声化学法制备了以聚甲基丙烯酸甲酯为壁材、以正十八烷为芯材的相变储能微胶囊,并对其进行粒径、扫描电镜、热失重、差示扫描量热及X射线衍射分析,主要结论如下:
(1)采用直接声化学法合成出了相变储能微胶囊,其粒径分布较为均匀,约为540 nm。
(2)采用乳化后再进行声化学反应合成的相变储能微胶囊,粒径大于直接声化学法合成的微胶囊,粒径约为650 nm。
(3)扫描电镜结果显示,相变储能微胶囊均呈现出比较规则的球形,且表面光滑。
(4)DSC结果显示,相变储能微胶囊的相变焓值低于纯十八烷,为178.9 J/g。
(5)Tg结果显示,壁材能够延缓正十八烷的分解,从而提高微胶囊的热稳定性。
(6)XRD结果显示,相变储能微胶囊的特征峰与正十八烷特征峰相比有所不同,间接证明了聚甲基丙烯酸甲酯壁材对正十八烷芯材的包覆。
