Transition-metal-catalyzed switchable divergent cycloaddition of para-quinone methides and vinylethylene carbonates: Access to different sized medium-sized heterocycles
2022-11-05JunweiWngLeiZhoChenZhuBenXiolongXieJinLiuShiyunHeMgnusRuepingKunZhoLihongHu
Junwei Wng,Lei Zho,Chen Zhu,Ben M,Xiolong Xie,Jin Liu,Shiyun He,Mgnus Rueping,Kun Zho,Lihong Hu,*
a Jiangsu Key Laboratory for Functional Substances of Chinese Medicine,School of Pharmacy,Nanjing University of Chinese Medicine,Nanjing 210023,China
b KAUST Catalysis Center (KCC),King Abdullah University of Science and Technology (KAUST),Thuwal 23955-6900,Saudi Arabia
c Department of Medicinal Chemistry,Key Laboratory of Chemical Biology,School of Pharmacy,Cheeloo College of Medicine,Shandong University,Ji’nan 250012,China
Keywords:Medium-sized rings Divergent cycloaddition Regioselectivity para-Quinone methides Vinylethylene carbonates
ABSTRACT Divergent synthesis of medium-sized rings with controllable ring sizes represents a longstanding challenge in organic synthesis.Herein,we developed a transition-metal-catalyzed switchable divergent cycloaddition of para-quinone methides and vinylethylene carbonates by controlling the steric hindrance of substituent.Different from reported alkoxide-triggered annulations,this process undergoes a regiodivergent allylation of para-quinone methides followed by 1,6-addition reaction,providing a new route to selectively synthesize seven-to ten-membered nitrogen-containing heterocycles in high yields with excellent regioselectivities.This protocol features a broad substrate scope,wide functional group tolerance as well as operational simplicity.The reaction mechanism was investigated by conducting a series of control experiments as well as DFT calculations and the origins of the regioselectivities of the cycloaddition process were rationalized.
Medium-sized rings (MSRs,7-11 members),in particular Nand O-containing heterocycles,frequently appear in a wide range of bioactive natural products and pharmaceuticals [1,2].Among them,benzo-fused medium-ring heterocycles have gained increasing attention recently because of their potential pharmaceutical values (Scheme 1a) [3-5].However,the efficient construction of medium-sized heterocycles presents a formidable challenge in organic synthesis due to their inherent entropic factors and unfavorable transannular interactions [6].Although a number of synthetic strategies have been reported to prepare medium-sized rings,including ring-closing metathesis [7-9],metal-and organo-catalyzed cyclization reactions [10-13]and so on [14,15].Most established methods are only suitable for the synthesis of certain sized heterocycles,changing the size of the ring usually requires designing new methods and substrates,which is quite costly and inefficient.Therefore,it is highly desirable to develop a unified approach for divergent synthesis of medium-sized heterocycles with various ring sizes.To date,no divergent [4 +n]cycloaddition reactions (n= 3-6) have yet been documented.To fill this gap,we sought to develop a highly efficient divergent cycloaddition reaction to selectively construct seven-to ten-membered heterocycles.
Over the past decade,vinylethylene carbonates (VECs) have arisen as versatile precursors for Pd-containing dipole synthons in organic synthesis [16-18].Typically,in the presence of palladium catalysts,VECs could generate zwitterionicπ-allyl palladium species or palladacyclic intermediatesviaa decarboxylation process (Scheme 1b).The research groups of Kleij,Zhang and others have demonstrated that a wide range of nucleophiles could attack the terminal (C5) [19-26]or internal (C3) [27-29]position of Pd-π-allyl species to deliver linear or branched building blocks in high efficiency.Furthermore,applications of VECs in [3 + 2]or[5 +n]cycloaddition reactions to construct five-membered heterocycles [30-35]or MSRs [35-40]have also been well studied.Recently,Zhao and co-workers found that VECs could be used as a reaction switch for divergent cycloadditions.In 2017,they disclosed the first [5 + 4]/[3 + 2]cycloaddition of VECs and azadienes,affording benzofuran-fused nine-membered heterocycles and five-membered spirocyclic products,where VECs were served as“5” or “3” synthon and the steric hindrance played an important role in the switch of the regioselectivity [41].Quite recently,they reported a divergent [4 + 2]/[3 + 2]cycloadditions of VECs and azadienes,and an unprecedented switch from alkoxide-π-allyl to dienolate reactivity was achieved by the use of palladium-titanium relay catalysis in this case [42].
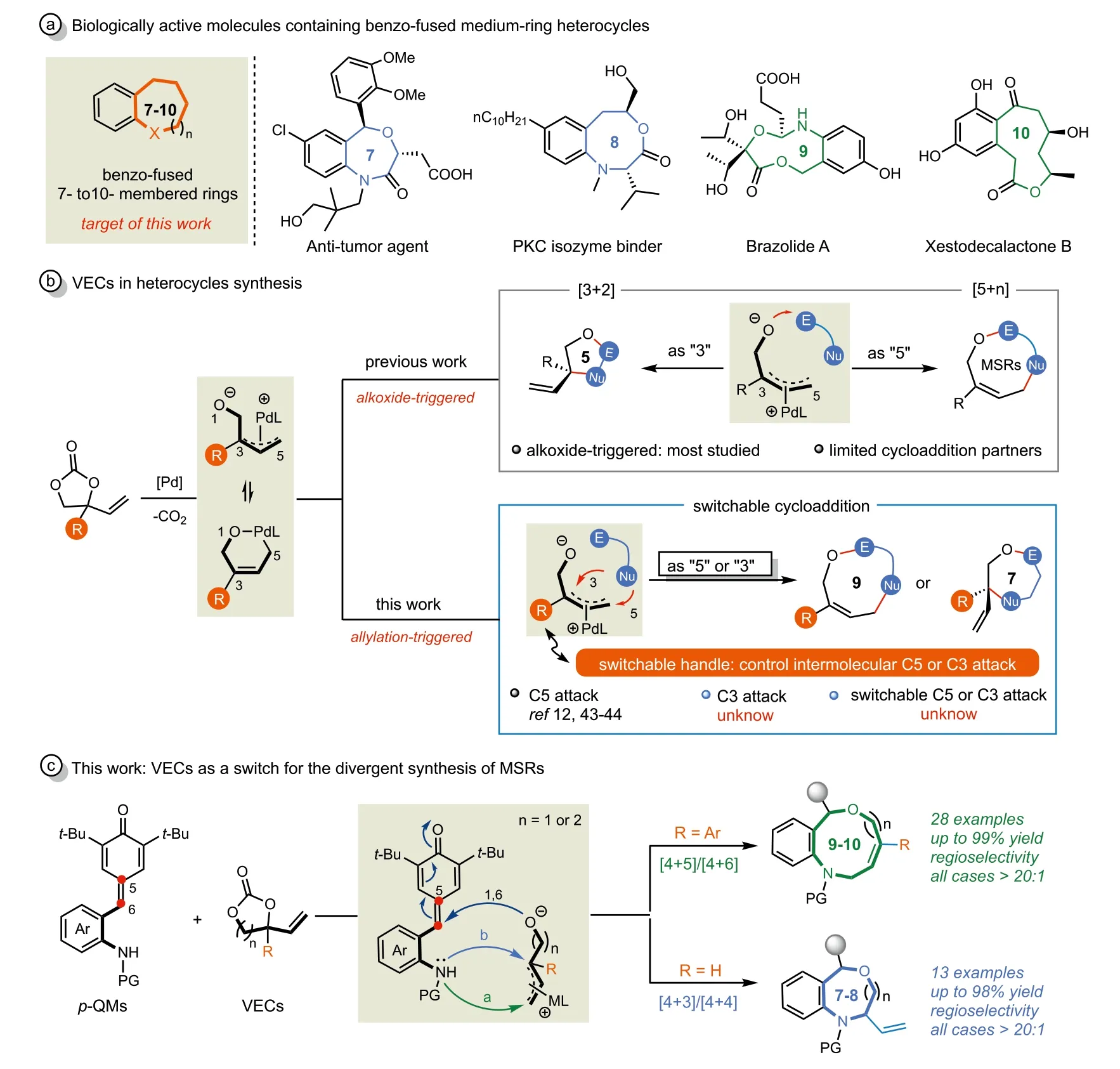
Scheme 1.Profile of VECs-involved cycloaddition reactions and our synthetic strategy.
Despite the progress made,most cycloaddition reactions of VECs were based on an alkoxide-triggered annulation process(Scheme 1b,top),the intermolecular allylation-triggered annulations have been less explored (Scheme 1b,bottom) [12,43,44].To the best of our knowledge,reactions relied on allylation-triggered processes,in which nucleophiles selectively reacted with C3 or C5 ofπ-allyl species followed by intramolecular annulations,are still unknown to date (Scheme 1b,bottom).Therefore,further application of VECs in novel reactions beyond oxygen anions-triggered cycloadditions for the construction of different sized MSRs skeleton is highly warranted.
Stimulated by pioneering studies of VECs and our ongoing research efforts onpara-quinone methides (p-QMs) [45-54],we were attracted to the cycloaddition ofortho-tosylaminophenylsubstitutedp-QMs and VECs.We envisioned that the annulation reaction betweenp-QMs and VECs should proceed in a cascade fashion,in which the sulfonamide motif would firstly attack on the terminal or internal position of Pd-π-allyl intermediates in a controllable manner followed by an intramolecular 1,6-addition reaction to deliver the [4 + 5]or [4 + 3]cycloaddition adducts respectively (Scheme 1c).
To test the feasibility of this intriguing hypothesis,we chosep-QM 1a and phenyl VEC 2a as the model substrates to optimize the reaction conditions (Table 1).Initially,we carried out the screening of different bases in the presence of Pd(PPh3)4(entries 1-6).First,several commonly used organic bases were evaluated,but the results were disappointing (entries 1-4).To our delight when DBU was employed,the expected reaction occurred,affording desired [4 + 5]adduct 3a in 66% yield with high regioselectivity,and no [4 + 3]adduct 4a was observed (entries 5 and 6).Encouraged by this result,another palladium precursor and more phosphine ligands were screened (entries 7-10),and it turned out that the combination of Pd2(dba)3·CHCl3and dppe ligand significantly improved the efficiency of this reaction (91% yield,entry 10).Next,the influence of solvent was examined,however,other solvents(e.g.,DCE,CHCl3,THF,acetone and acetonitrile) failed to give better results (entries 11-15).With the optimal solvent,the reaction temperature was further examined (entries 16-18) and we found that 60 °C was the appropriate temperature.After establishing the optimal base,catalyst and solvent,other reaction parameters such as the catalyst/ligand loading were finally examined (entries 19-21).We were pleased to find that a simple cocktail containing Pd2(dba)3·CHCl3(5 mol%),dppe (10 mol%),and DBU in toluene at 60 °C served as the best reaction conditions,and the expected product 3a could be isolated in an excellent yield (95%,entry 19).
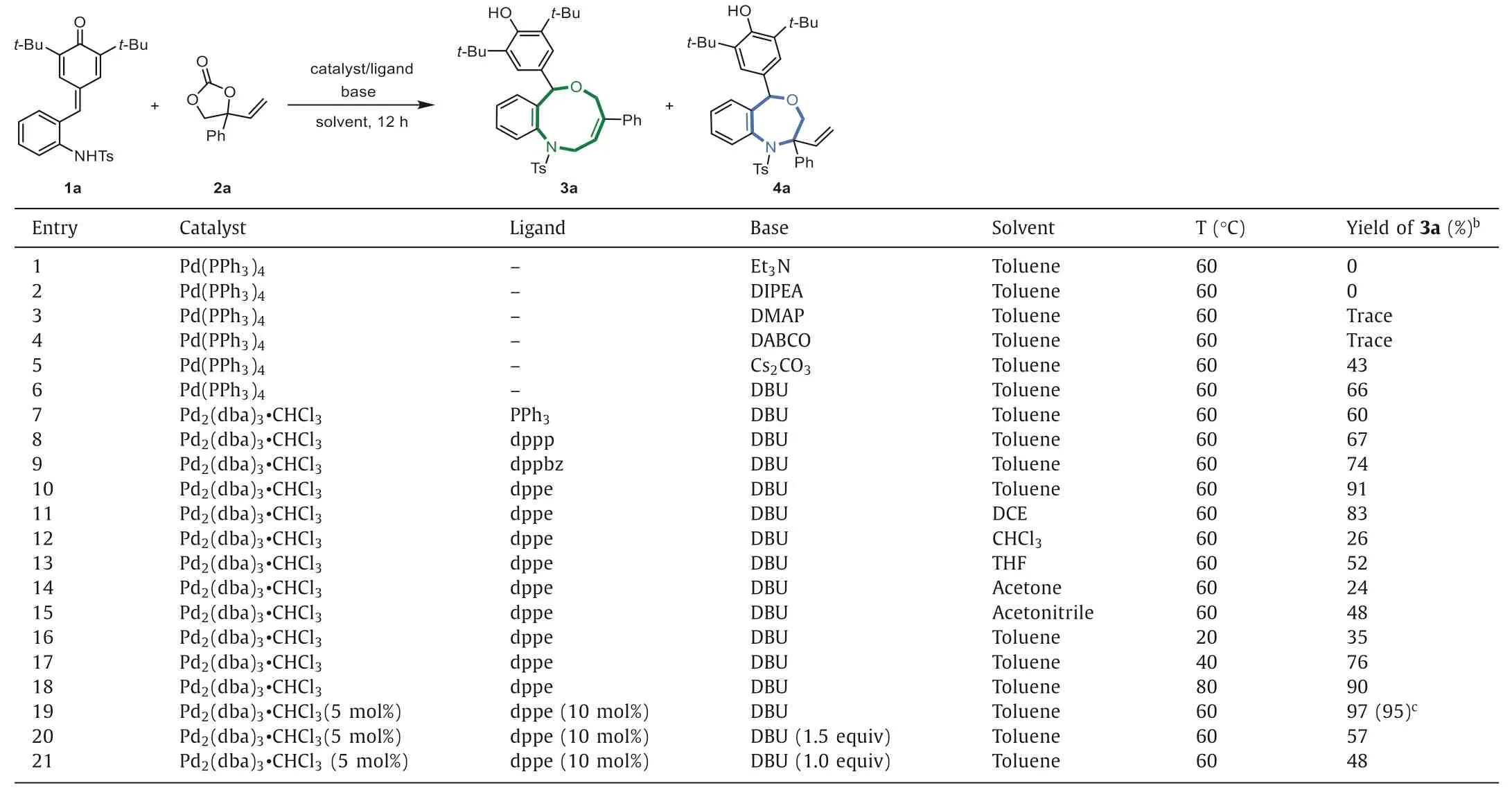
Table 1 Optimization of the reaction conditions.a
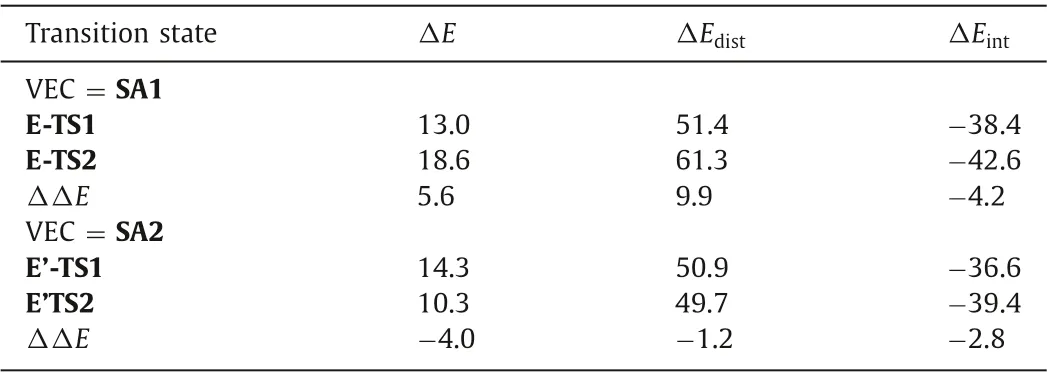
Table 2 Single point energies,distortion energies,and interaction energies for distortion/interaction analysis.Single point energies (in kcal/mol) at the SMD(toluene)-M06/Def2-TZVPP level are displayed.
After establishing the optimized conditions for the synthesis of nine-membered rings,we began to evaluate the generality of this protocol.First,the substrate scope ofp-QMs part was explored (Scheme 2,top).It was found thatp-QMs bearing different substituents on the benzene ring could be smoothly converted into the expected nine-membered heterocycles 3a-3i in high yields(74%-99%) and regioselectivities,and the reaction efficiency was less affected by the electronic properties or the position of substituents.The benzene sulfonyl protective group on the nitrogen atom ofp-QMs also did not affect the efficiency of this reaction,and the corresponding products 3j-3l were obtained in 81%-90%yields.When the benzene sulfonyl protective group was replaced with the methyl sulfonyl protective group,the yield of 3m was reduced to 47%.Subsequently,the scope of VECs 2 was investigated by varying the substituents on the benzene ring or the size of the R group of phenyl VECs (Scheme 2,bottom).We were pleased to find that variouspara-,meta-,orortho-substituents with both electron-rich and electron-poor character on the benzene ring of VECs were well-tolerated,and the corresponding products 3n-3v were delivered in excellent yields (70%-99%).Moreover naphthyland heteroarene-substituted VECs could also readily take part in this [4 + 5]annulation reaction,affording desired products 3w and 3x in 76% and 57% yield,respectively.
Notably,when aryl groups were replaced by ethylene or alkyl substituents,the corresponding products 3y-3aa were obtained in lower yields (9%-34%).Those results indicated that the reaction efficiency was influenced by the substituent of VECs.We decided to further decrease the steric hindrance of the substituent.Non-bulky substituted-VEC 2a’(R = H)was synthesized and exposed to the optimal conditions.To our surprise,the nine-membered product 3ab was not formed and a seven-membered ring product 5a was isolated,albeit with low yield (20%).In this case the regioselectivity of this annulation process was completely switched,and the sulfonamide species firstly attacked the C3 position of the Pd-πallyl intermediate forming a kinetically favored seven-memberedring structure.
Striving for higher efficiency of this [4 + 3]cycloaddition reaction,an optimization study was carried out (see Supporting information for optimization details).We evaluated a range of phosphine ligands,metal catalysts as well as several bases,among which the combination of [Ir(cod)Cl]2and DABCO was proved to be the optimal reaction choice.Under this condition,[4 + 3]cycloaddition adduct 5a was isolated in an excellent yield (95%) with high diastereoselectivity (>20:1).With the optimal conditions in hand,the generality of this [4 + 3]cycloaddition reaction was further explored.As shown in Scheme 3,the substrate scope of this [4 + 3]annulation proved to be very broad as well,a wide range ofp-QMs 1 bearing electron-rich or electron-poor groups at different position of the benzene ring readily reacted with VEC 2a’to afford expected seven-membered ring products 5a-5h in high yields and diastereoselectivity.In addition,p-QMs 1 with differentN-protecting groups were also suitable substrates for this transformation,giving corresponding products 5i-5l in 83%-98% yields.The structure and relative configuration of 3a and 5a were unambiguously determined by the single-crystal X-ray diffraction analysis,and the structures of other nine-and seven-membered products were assigned by analogy.
To evaluate the robustness and practicality of this novel protocol.A scale-up reaction was performed under the standard conditions.As shown in Scheme 4a,both [4 + 5]and [4 + 3]annulation reactions worked very well on 1 mmol scale,and the corresponding products 3a and 5a could be isolated in 93% and 94% yields,respectively.To illustrate the versatility of products,we turned our attention to the exploration of selective transformation of those cycloadditions adducts.To our delight,in the presence of PtO2catalytic hydrogenation of the olefin moiety of 3a afforded hydrogenated product 6 in 55% yield.Treating withm-CPBA diastereoselective epoxidation of 3a yielded epoxide 7 in a good yield (73%)with high diastereoselectivity (>20:1) (Scheme 4b,top).The Heck reaction of 5a with idobenzene gave styrene derivative 8 in satisfied results (71% yield;>20:1 dr).Seven-membered adduct 5a could undergo olefin metathesis in the presence of Grubbs II catalyst,giving the corresponding product 9 with an acceptable yield and good diastereoselectivity (Scheme 4b,bottom).
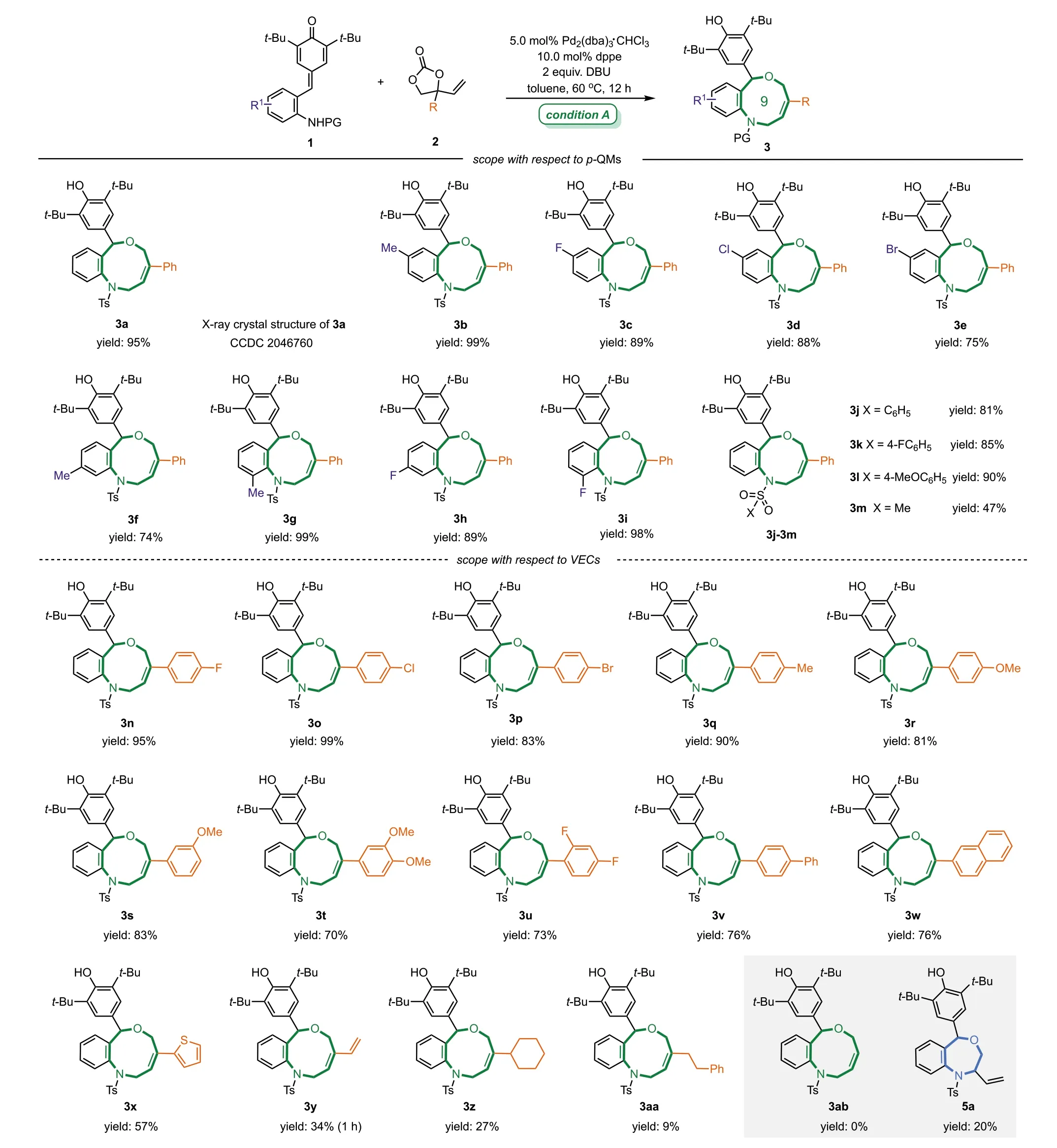
Scheme 2.Substrate scope of the [4 + 5]cycloaddition reaction.Unless noted otherwise,the reaction was performed under condition A: 1 (0.10 mmol),2 (0.15 mmol),Pd2(dba)3·CHCl3 (5.0 mol%),dppe (10.0 mol%),DBU (2.0 equiv.),toluene (1.5 mL),60 °C,12 h.Yields of isolated products.
Next,we turned to explore the enantioselective version of this reaction.As demonstrated in Scheme 5a,a large number of common chiral phosphine ligands were screened,however,only a moderate enantioselectivity was achieved and compound 3a was isolated with 73:27 er (see Supporting information for more details).In addition,we extended this divergent cycloaddition strategy to the reaction between 1a and six-membered vinyl carbonate 10,assembling ten-and eight-membered heterocycles 11 and 12 in satisfying yields and excellent regioselectivity (Scheme 5b).The structure of the ten-membered heterocycle 11 was unambiguously assigned by the single-crystal X-ray diffraction analysis.
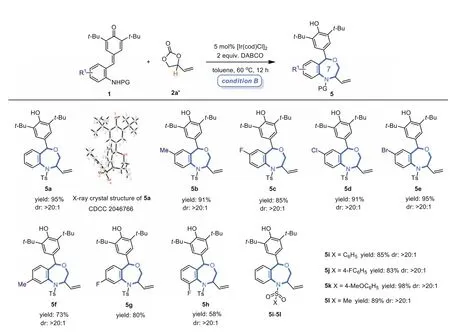
Scheme 3.Substrate scope of the [4 + 3]cycloaddition reaction.Unless noted otherwise,the reaction was performed under condition B: 1 (0.10 mmol),2a’(0.15 mmol),[Ir(cod)Cl]2 (5.0 mol%),DABCO (2.0 equiv.),toluene (1.5 mL),60 °C,12 h.Yields of isolated products.
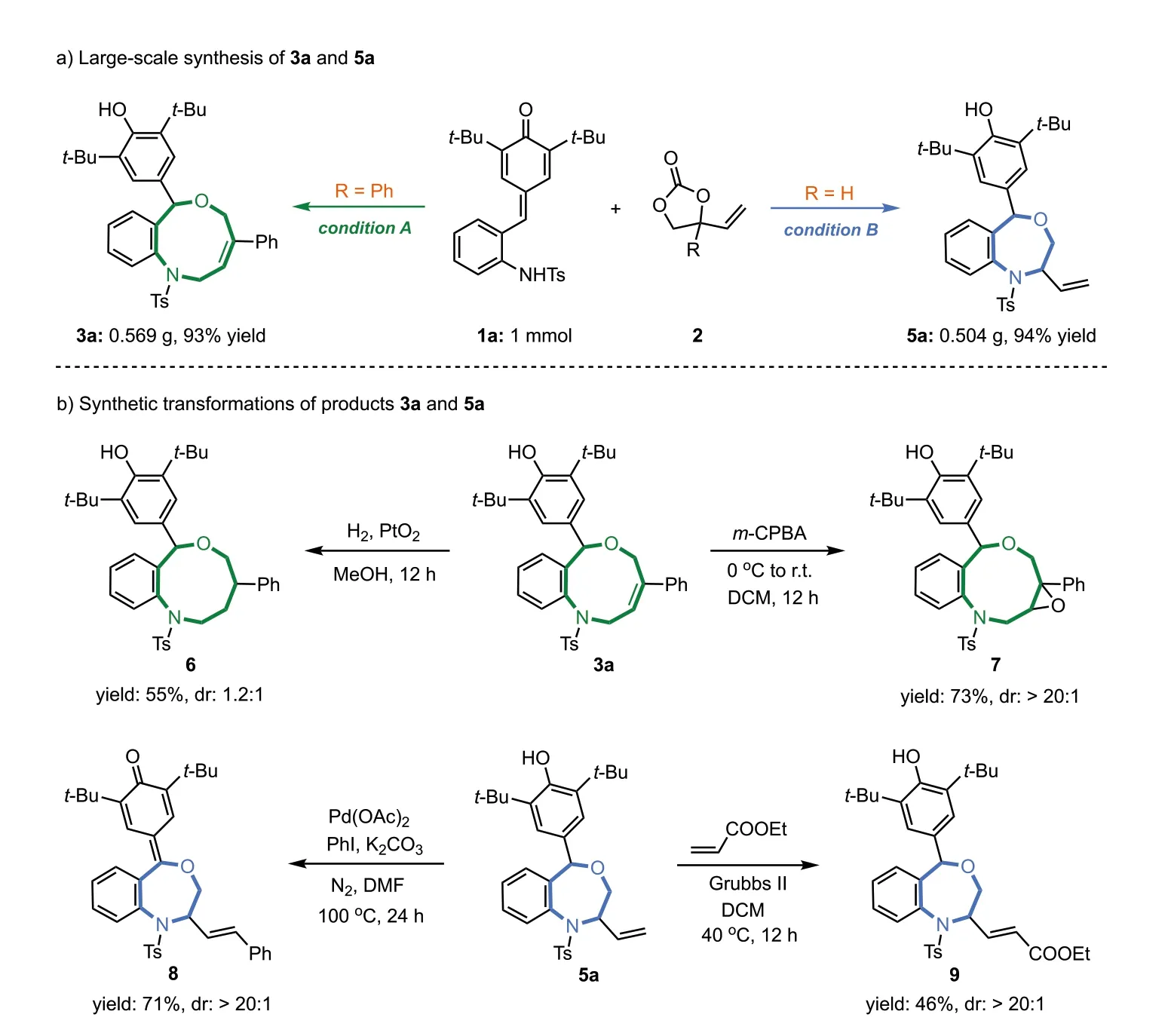
Scheme 4.Scale-up preparation and synthetic applications.
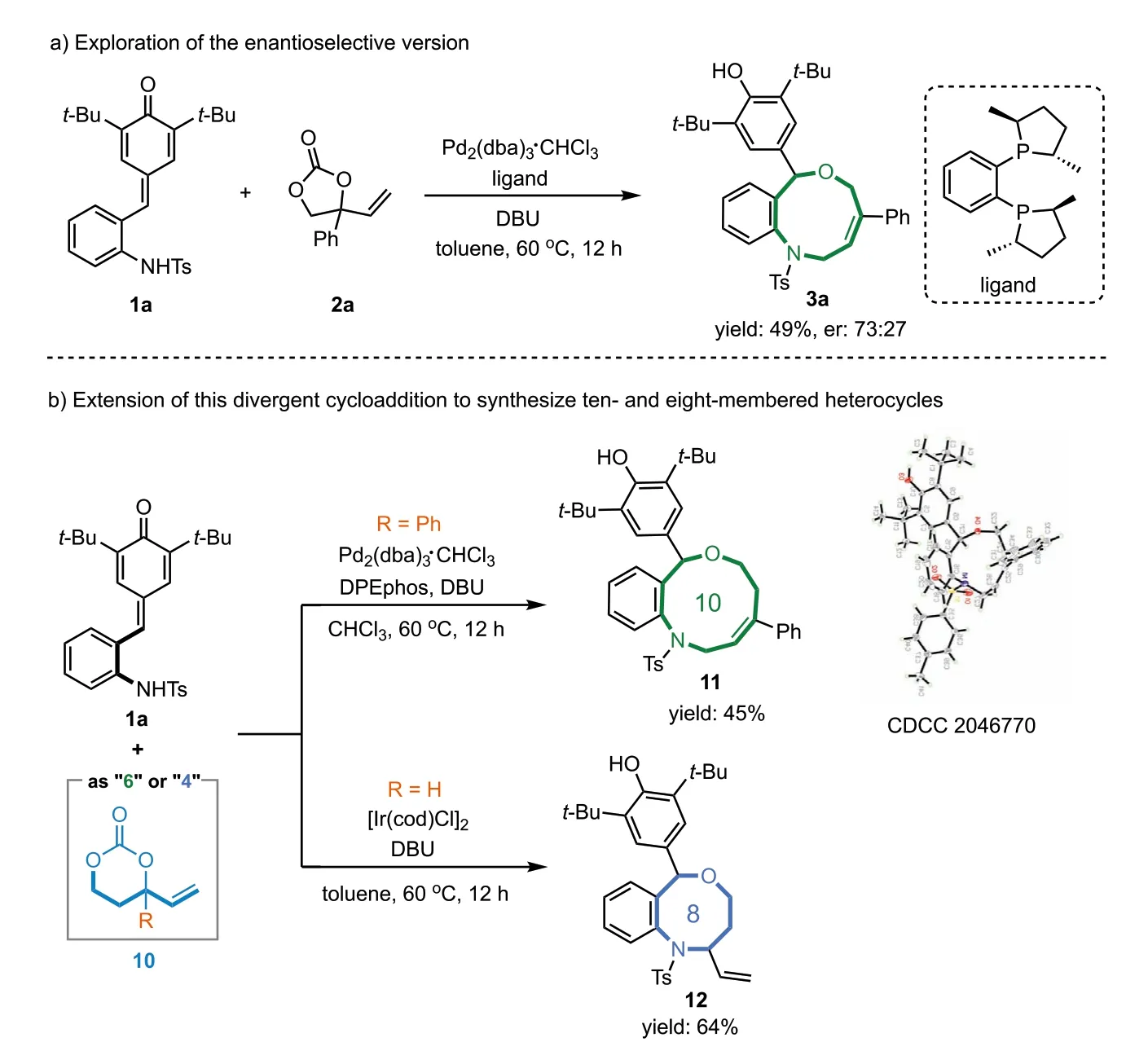
Scheme 5.Extension of this divergent cycloaddition.
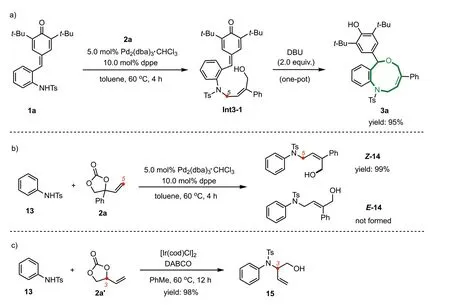
Scheme 6.Control experiments.
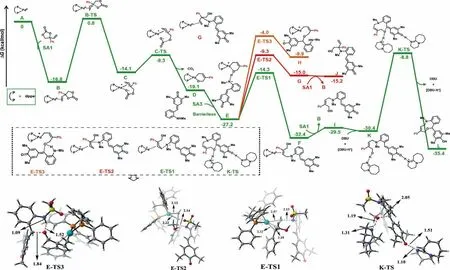
Fig.1.DFT-computed energy profiles for the [4 + 5]cycloaddition of SA3 and SA1.Free energies in solution (in kcal/mol) at the SMD(toluene)-M06/Def2-TZVPP//PBE/SDD(Pd)/Def2-SVP (other atoms) level are displayed.DFT optimized geometries of selected transition states are shown.Bond lengths are in ˚A.
Besides the substrate scope of this reaction,we were also interested in the mechanism of this switchable process.In order to probe the reaction pathway,some preliminary control experiments were conducted.As shown in Scheme 6a,the reaction ofp-QM 1a with phenyl VEC 2a under standard conditions (without DBU) was carried out,and we obtained an uncyclic compound Int3-1 rather than the cyclic product 3a.After subsequently adding DBU,the final product 3a could be afforded in high yield (95%).This result indicated that the overall process was trigged by an intermolecular alkylation of sulfonamides,and then an intramolecular 1,6-conjugate addition reaction occurred to deliver the final cycloaddition adducts.In order to study selectivity issues (terminal versus internal attack,E-versus Z-selectivity),we performed a reaction between simplified substrate 13 and phenyl VEC 2a under standard conditions,and the terminal-attacked product 11 was isolated in high yield as well as with excellentZ-selectivity (Scheme 6b);in contrast when the unsubstituted-VEC 2a’was used,a high C3-selectivity was achieved and the corresponding branched product 15 was obtained in quantitative yield (Scheme 6c).
Finishing those control experiments,a computational investigation by density functional theory (DFT) was undertaken to better understand the details of the whole process,specifically elucidating the origin of the observed regioselectivity of the reaction.All calculations were performed at the SMD(toluene)-M06/Def2-TZVPP//PBE/SDD(Pd)/Def2-SVP (other atoms) level of theory[55-62],and the computational details are given in Supporting information.First,a simplified variant ofp-QMs SA3 (t-Bu and Ts groups were replaced with Me groups) and phenyl substituted VEC SA1 were chosen as model substrates to study the energy profile for the [4 + 5]cycloaddition reaction (Fig.1).The reaction starts with the oxidative cleavage of phenyl substituted VEC SA1 by the active catalyst,Pd0(dppe) (A),which proceedsviaadduct B and transition state B-TS.This cleavage step has an energy barrier of 17.6 kcal/mol.The resulting intermediate C has anη3-allylic group with an open carbonate.Next,a facile decarboxylation of the open carbonate from the intermediate C gives the six-membered palladacyclic intermediate Dviathe transition state C-TS with only 4.8 kcal/mol energy barrier.In the next step,thep-QM SA3 transfers a proton to the anionic oxygen center of D.This process is barrierless,and the generated intermediate E is 8.1 kcal/mol more stable.Starting from the intermediate E,the energy barriers of three possible nucleophilic attacks were calculated.Both the terminal C5 position and internal C3 position can be nucleophilically attacked by the anionic nitrogen from SA3.The attack on the terminal C5 position has the lowest energy barrier of 12.9 kcal/mol (transition state E-TS1),while the barrier for the internal C3 position attack is 5 kcal/mol higher (transition state E-TS2).The regioselectivity is determined from this terminal nucleophilic attack step.In addition,the third reaction possibility that the nucleophilic attack of the anionic oxygen on thep-QM SA3 was also proved to be less favored,with the highest activation barrier of 23.2 kcal/mol.This result is consistent with the experimental result (Scheme 6a).To push the reaction forward,in the presence of DBU the hydroxyl group of intermediate I could be deprotonated,followed by the intramolecular 1,6-conjugate addition with an energy barrier of 21.6 kcal/mol (transition state K-TS).The final nine-membered heterocycle product L is formed through this reaction pathway.
We also conducted the DFT calculations for the [4 + 3]cycloaddition reaction between a simplifiedp-QM SA3 and unsubstituted VEC SA2 (Fig.2).Similar to phenyl substituted VEC SA1,the cyclic carbonate SA2 undergoes oxidative cleavage and CO2 extrusion to form the intermediate D’,with the energy barriers of 17.7 kcal/mol and 5.1 kcal/mol,respectively.After thep-QM SA3 transfers the proton to the anionic oxygen center of D’,the nucleophilic nitrogen from SA3 can attack the C5 or C3 position of theη3-Pd-allyl species.In contrast to the energy profile in Fig.1,the attack on the internal C3 position has the lowest energy barrier of 11.1 kcal/mol when unsubstituted VEC SA2 was used here.The nucleophilic attack on the terminal carbon (C5 position) has a higher activation barrier (13.6 kcal/mol),which is kinetically unfavored.Next,the DBU promotes the intramolecular 1,6-conjugate addition reaction with the energy barrier of 17.6 kcal/mol and gives the seven-membered heterocycle product L’.The DFT calculations demonstrated that the regioselectivity was determined by the nucleophilic attack step (E to F and E’to G’).When phenyl substituted VEC SA1 was used,the attack on the terminal position of theη3-allylic group was kinetically favored.In contrast,when unsubstituted VEC SA2 was employed,the attack on the internal position has the lowest energy barrier.These DFT computational results were in accordance with experimentally observed regioselectivity of those switchable cycloadditions of 1a and 2.
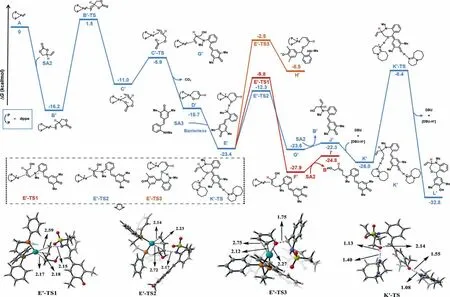
Fig.2.DFT-computed energy profiles for the [4 + 3]cycloaddition of SA3 and VEC SA1.Free energies in solution (in kcal/mol) at the SMD(toluene)-M06/Def2-TZVPP//PBE/SDD(Pd)/Def2-SVP (other atoms) level are displayed.DFT optimized geometries of selected transition states are shown.Bond lengths are in ˚A.
To gain a clearer understanding of the origin of the regioselectivity controlled by the VEC subrates (SA1 and SA2),we performed the distortion/interaction analysis [63]of the nucleophilic attack transition states (Table 2).The contributions of distortion and interaction were calculated as follows:

Our analysis indicated that the distortion energy is mostly responsible for the regioselectivity.For both VECs,the internal C3 position attacks have a higher interaction energy(ΔΔEint=-4.2 kcal/mol and-2.8 kcal/mol,respectively).However,when phenyl substituted VEC SA1 is employed,the internal C3 position attack (E-TS2) has a significantly greater distortion energy (61.3 kcal/mol),which could be a result of the phenyl group’s steric effect on the SA1 (see structures of E-TS1 and E-TS2 in Fig.1).
Based on our preliminarily control experiments and computational results,we proposed a plausible mechanism that is same to the description of the computational study (Fig.3).
In summary,we have successfully developed a regiodivergent cycloaddition of VECs withp-QMs.This transformation provides a highly selective protocol to rapidly access a wide range of mediumsized heterocycles.The substituents of VECs are found to be critical to obtain high regioselectivity,and by changing the steric properties of the substituents we could achieve either [4 + 5]or [4 + 3]cycloaddition reactions,affording nine-or seven-membered benzofused heterocycles which cannot be readily synthesized using the previous methods.In addition,this protocol is characterized by a broad substrate scope and excellent regioselectivity.Moreover,this method could be extended to the synthesis of eight-and tenmembered heterocycles.Control experiments supported that this switchable cycloaddition reaction proceeds in a domino fashion,the triggered step involves an intermolecularN-allylic substitution,followed by 1,6-conjugate addition reaction.The DFT calculation revealed that the steric hindrance of the substituent on vinyl carbamates is the key factor that account for the regioselectivity.
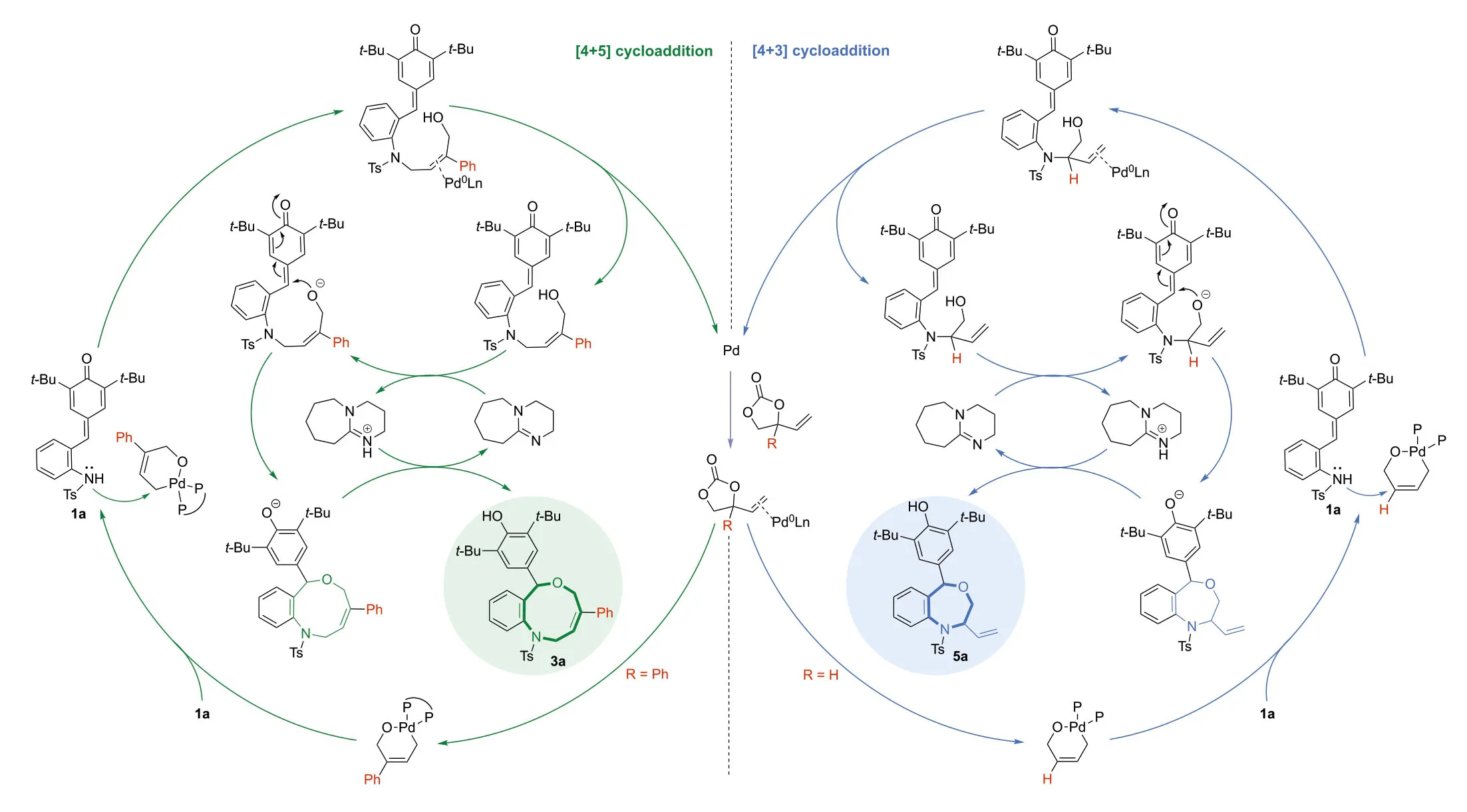
Fig.3.Proposed mechanism.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos.82173664,81803342),“ShuangChuang” Research Team of Jiangsu Province (No.20182036).C.Zhu acknowledges King Abdullah University of Science and Technology (KAUST)for support and the KAUST Supercomputing Laboratory for providing computational resources of the supercomputer Shaheen II.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.01.063.
杂志排行

Chinese Chemical Letters的其它文章
- An odyssey of lithium metal anode in liquid lithium-sulfur batteries
- Recent progress on preparation and applications of layered double hydroxides
- Two-dimensional transition metal chalcogenide nanomaterials for cancer diagnosis and treatment
- Emerging nanomedicine and prodrug delivery strategies for the treatment of inflammatory bowel disease
- Recent advances in persulfate-based advanced oxidation processes for organic wastewater treatment
- Recent advance of fluorescent probes for detection of drug-induced liver injury markers