普通小麦颖壳蜡质缺失突变体glossy1的转录组分析
2022-11-05李玲红陈永明尤明山倪中福邢界文
李玲红 张 哲 陈永明 尤明山 倪中福 邢界文
普通小麦颖壳蜡质缺失突变体的转录组分析
李玲红 张 哲 陈永明 尤明山 倪中福 邢界文*
中国农业大学农学院, 北京 100193
为了明确突变体颖壳蜡质含量显著变化的分子机制, 本研究对源自济麦22颖壳蜡质缺失突变体与野生型进行了转录组分析。结果表明, 在突变体中, 共筛选到12,230个差异表达基因, 其中5811个基因在突变体中上调表达, 6419个下调表达。GO (gene ontology)功能富集分析发现, 差异基因主要富集在蜡质合成和转运途径, 具体分布在酰基转移酶活性、脂质结合和水解酶活性等条目, 由此推测这些途径与小麦穗部蜡质缺失性状是紧密相关的。我们还利用RT-qPCR检测了参与蜡质代谢途径部分基因的表达, 结果与转录组结果是一致的。本研究为今后探究小麦蜡质代谢的分子机制和基因调控网络提供了数据支持, 同时也为抗逆小麦育种奠定了理论基础。
小麦; 表皮蜡质;突变体; 转录组分析; 分子机制
小麦是全球范围内广泛种植的重要粮食作物,总产量仅次于玉米。表皮蜡质(Epicuticular wax)是指覆盖在植物地上部分表面的一层脂质有机混合物, 主要由烷烃类、醇类、醛类、酮类和脂肪酸等化合物组成[1]。表皮蜡质是植物与环境之间的保护屏障, 能够保护植物免受干旱、紫外线、病原菌和昆虫等的侵害[2]。因此, 研究表皮蜡质相关基因及调控机制对培育抗逆性作物品种, 提高粮食产量具有重要意义。
植物中蜡质化合物的合成发生在表皮细胞内, 主要分为3个阶段。第一, 在质体(Plastid)中合成C16和C18脂肪酸: 以乙酰辅酶A为底物, 通过脂肪酸合酶(FAS)复合物对C2的连续缩合而伸长[3-4]; 第二, 在内质网(endoplasmic reticulum)中通过脂肪酸延伸酶复合物(FAE)进一步延伸为长链脂肪酸化合物(VLCFAs): 利用质体生成的C16或C18脂肪酸和丙二酰辅酶A (CoA)作为底物, 通过脂肪酸延伸酶(FAE)复合物, 在KCS、KCR、HCD和ECR四种酶的催化下循环反应生成C20~C34化合物[5]; 第三, 生成的VLCFAs经过不同的蜡质合成途径生成不同的化合物。
植物中普遍存在两条蜡质生物合成途径, 包括烷烃合成途径(也叫脱羰途径)和醇合成途径(也叫酰基还原途径)。前者通过CER1、CER3和CYTB5复合物催化由醛生成烷烃, 继而通过中链烷烃羟化酶1 (MAH1)生成仲醇和酮[6]; 后者通过脂肪酸酰基辅酶A还原酶(CER4/FAR3)和蜡合酶/二酰基甘油酰基转移酶(WSD1)分别产生初级醇和蜡酯[7-8]。小麦不同于其他植物, 在生殖生长阶段有一条独特的蜡质生物合成途径: β-二酮途径[9]。β-二酮主要为正十三烷-14,16-二酮, 由于β-二酮的存在, 绝大多数现代小麦(L.)品种表现出白霜状蜡质外观[10-11]。近期研究证实小麦中负责β-二酮的生物合成的位点是一个合成基因簇, 分别编码III型的聚酮合成酶、一个细胞色素P450和一个水解酶/羧化酯酶[12]。不同蜡质化合物从内质网合成以后转移到质膜(PM), 然后由ABCG转运蛋白穿过PM, 最后脂质转运蛋白(LTP)参与穿过外周细胞壁转移到角质层表面, 自我组装成肉眼可见的白霜状表皮蜡质层[13-14]。
前期我们利用甲基磺酸乙酯(EMS)诱变小麦品种济麦22获得一个颖壳蜡质缺失突变体, 命名为突变体, 并对其进行了表型鉴定、遗传分析和精细定位。GC-MS测定结果显示, 与野生型相比, 突变体的总蜡质含量和单个蜡组分含量均发生显著变化, 特别是突变体中β-二酮含量显著高于野生型[15]。为了阐明潜在的分子机制, 本文对济麦22和抽穗期的颖壳进行了转录组比较分析。研究结果揭示了基因调控小麦蜡质合成代谢相关基因的可能的分子机制, 为构建蜡质代谢表达调控网络提供理论依据, 同时也为进一步选育抗逆小麦品种奠定基础。
1 材料与方法
1.1 试验材料
小麦突变体是由小麦优质栽培品种济麦22经EMS诱变所得, 经连续自交, 已纯合一致, 能稳定遗传。相较于野生型,突变体的穗部颖壳明显表现为光滑无蜡(图1)。2017年秋季, 在北京上庄(116°E, 40°N)分别种植济麦22和突变体, 并设置3个重复(3行/重复)。播种时行长1.5 m, 间距0.3 m, 籽粒均匀分布, 每行播种率为20粒。

图1 济麦22和glossy1突变体的表型比较
A: 济麦22和突变体的整株表型, 标尺为1 cm; B: 穗子放大图, 标尺为1 cm。
A: The whole plant of Jimai 22 and.Bar: 10 cm; B: Enlarged view of spikes of Jimai 22 and.Bar: 1 cm.
1.2 总RNA提取和测序
2018年夏季, 于小麦开花期(GS65)收集野生型和突变体小麦颖壳组织, 并迅速置于液氮罐中, 作为后续转录组测序的样品。本研究对6个样品进行分析, 分别是突变体的3个生物学重复(命名为Rep1、Rep2和Rep3), 野生型济麦22的3个生物学重复(Jimai 22 Rep1、Rep2和Rep3)。液氮研磨样品, 利用TRIzol试剂(Invitrogen)提取颖壳总RNA, 具体方法参照说明书。完成总RNA提取后, 组织样品送至北京贝瑞和康生物技术有限公司利用HiSeq2000仪器完成文库构建、质量控制及转录组测序。
1.3 差异表达基因的鉴定
测序得到的原始数据(Raw data), 去除接头序列和低质量测序片段之后, 获得高质量的序列(Clean data)。将Clean data与小麦参考基因组(Chinese Spring, RefSeq v.1.1)进行比对, 获取基因组位置信息。使用RSEM软件包评估基因表达水平。使用FPKM (Fragments Per Kilobase of exon model per Million mapped fragments)方法标准化基因的表达水平[16]。使用DESeq R package[17], 使用调整后的值(FDR < 0.05)和Fold Change (FC)比值(|log2FC|≥1)来确定突变体和野生型之间的差异表达基因(DEGs)。
1.4 RT-qPCR验证差异表达基因
根据样品基因表达量差异, 本实验随机挑选9个基因(;;;;;;;;)进行实时荧光定量PCR (RT-qPCR)验证。吸取1 µg总RNA使用反转录试剂盒HiScript II RT SuperMixqPCR (Vazyme)试剂盒合成cDNA第1链。以(TraesCS5D02G132200)作为内源性对照校准基因表达水平。RT-qPCR的引物对列于表1。qPCR使用SYBR Green PCR Master Mix (Vazyme Biotech, Ltd., China)和CFX96 Real-time PCR检测系统(Bio-Rad Laboratories, Inc., USA)进行。RT-qPCR反应条件为: 95℃预变性3 min, 95℃变性15 s、60℃退火15 s、72℃延伸30 s设置40个循环, 在72℃延伸30 s阶段采集荧光, 并添加溶解曲线, 结果用2–ΔΔCt计算基因表达差异[18]。本实验包含3次生物学重复, 每个生物学重复进行3次技术重复。
1.5 差异表达基因的注释
为进一步了解差异表达基因(DEGs)的生物学意义, 本研究使用FDR<0.05作为筛选标准对DEGs进行基因本体论(GO)富集分析[19]。具体分析流程使用中国农业大学小麦研究中心生信分析平台中小麦族同源基因注释数据库Triticeae-Gene Tribe板块中的GoEnrichment功能模块进行分析[20]。
2 结果与分析
2.1 转录组数据质控
为了阐明济麦22背景的突变体穗部蜡质缺失性状的分子基础, 我们选取两亲本开花期的颖壳为材料提取总RNA, 进行了转录组测序(RNA-seq)。每个基因型设置3个生物学重复, 剔除低质量的测序reads后, 将每个样本的高质量reads比对到小麦中国春参考基因组(RefSeq v.1.1)上。结果显示, 每个样本的测序原始数据分别从73.17 M到91.29 M不等。经过质量过滤后, 每个样本产生52.12 M到64.66 M干净读长, 这些读长在参考基因组的比对率均达70.80%以上(表2)。同组样品基因表达模式趋于一致且Pearson相关系数均大于0.80 (图2-A)。总的来说, 这些结果均验证了RNA和测序数据的高质量, 可以进行后续分析。

表1 RT-qPCR所用引物
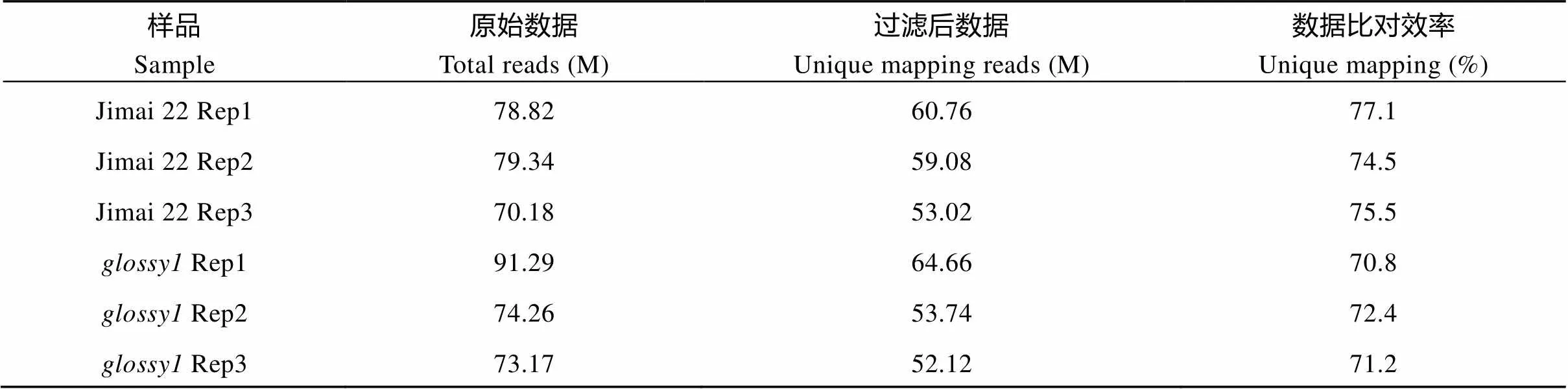
表2 样品测序输出数据的质量评价
2.2 差异表达基因分析
为了确定野生型和突变体之间基因表达的差异,根据FPKM值对基因表达水平进行标准化。所有的唯一映射读取被用来计算基因FPKM值。根据DESeq2分析结果, 以adjusted-value < 0.05且Fold-change value > 2 或者 < 0.5作为标准进行筛选, 共筛选出12,230个差异表达基因(DEGs), 占检测到基因总数的26.6%。相较于济麦22, 5811个基因(占总DEGs的47.5%)在突变体中上调表达, 6419个基因(占总DEGs的52.5%)在突变体中下调表达(图2-B)。进一步对12,230个差异表达基因的染色体分布进行了统计, 结果发现差异表达基因在A、B和D基因组上的分布基本是均匀的(图2-C)。
2.3 RT-qPCR验证
为了验证我们转录组数据的可靠性, 我们随机选择了9个差异表达基因进行实时荧光定量PCR的检测。实时定量PCR的结果与转录组数据的结果变化趋势比较一致(图3), 说明我们转录组数据有很高的可靠性。
2.4 差异表达基因GO富集分析
为了解济麦22和差异表达基因的生物学意义, 按照错误发现率(FDR)<0.05的标准对这些基因进行基因本体论(gene ontology, GO)富集分析。对12,230个差异表达基因进行GO功能注释和分析发现, 共有5380个基因获得注释, 分布于159个GO分类条目。根据基因数目分别筛选出30条最显著的条目(图4-A)。在三类注释生物学过程(biological process)、细胞组分(cellular component)和分子功能(molecular function)中所占的比例分别为46.5% (74)、8.2% (13)和45.3% (72)。在生物学过程中, 以抗氧化过程(response to oxidative stress)的基因数最多, 脂肪酸生物合成过程(fatty acid biosynthetic process)以及脂质转运(lipid transport)等生物学过程的基因数目占很大比例。分子功能注释的基因大部分集中在水解酶活性(hydrolase activity)、转移酶活性(transferase activity)、单加氧酶活性(monooxygenase activity)以及脂质结合(lipid binding)等。在细胞组分中, 质膜(plasma membrane)功能组拥有的基因数最多。
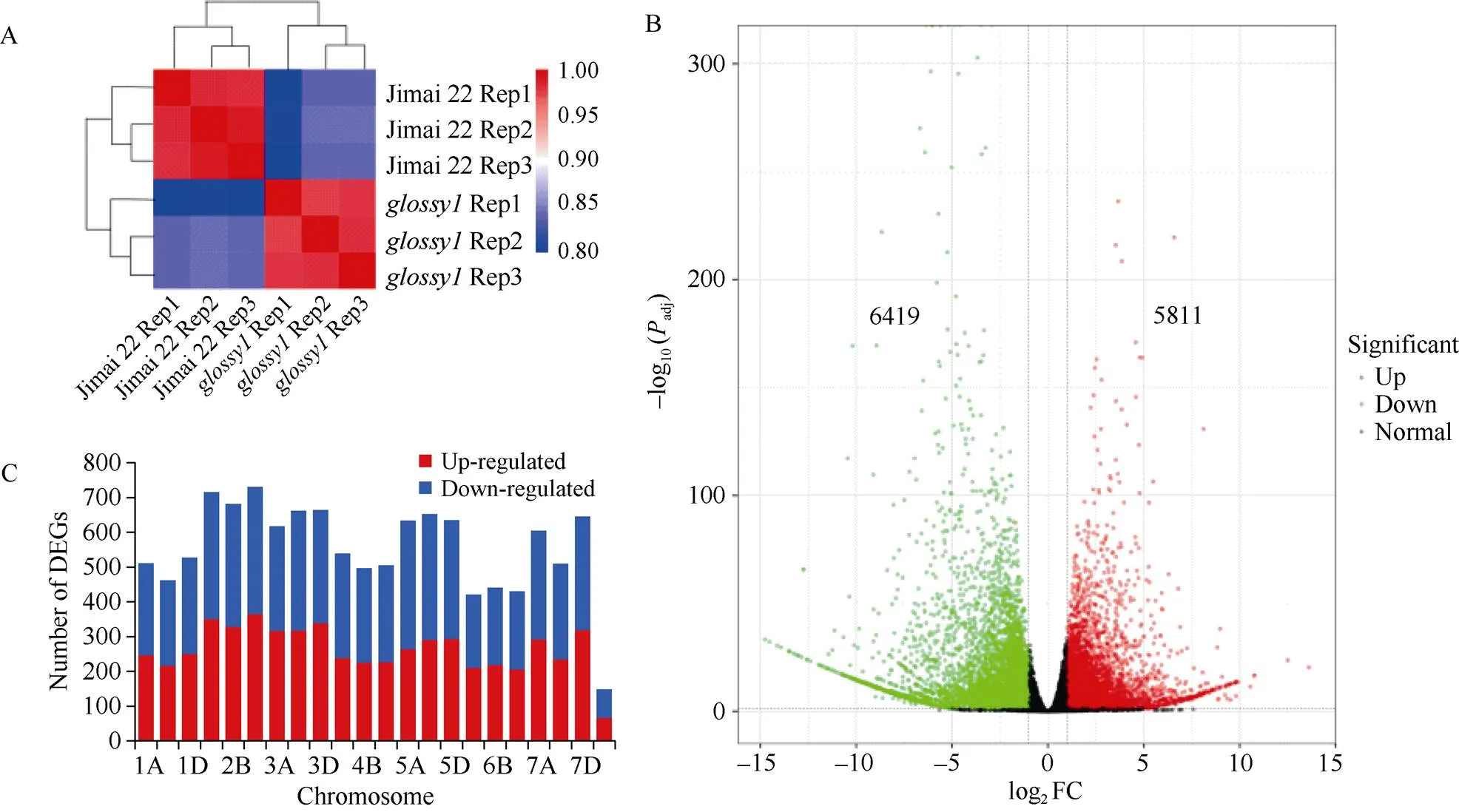
图2 样品相关性及差异表达基因分析
A: 样品相关性分析; B: 差异表达基因火山图; C: 差异表达基因在染色体上的分布。
A: correlation analysis among the samples; B: volcano plot of DEGs; C: the distribution of DEGs on wheat chromosomes.
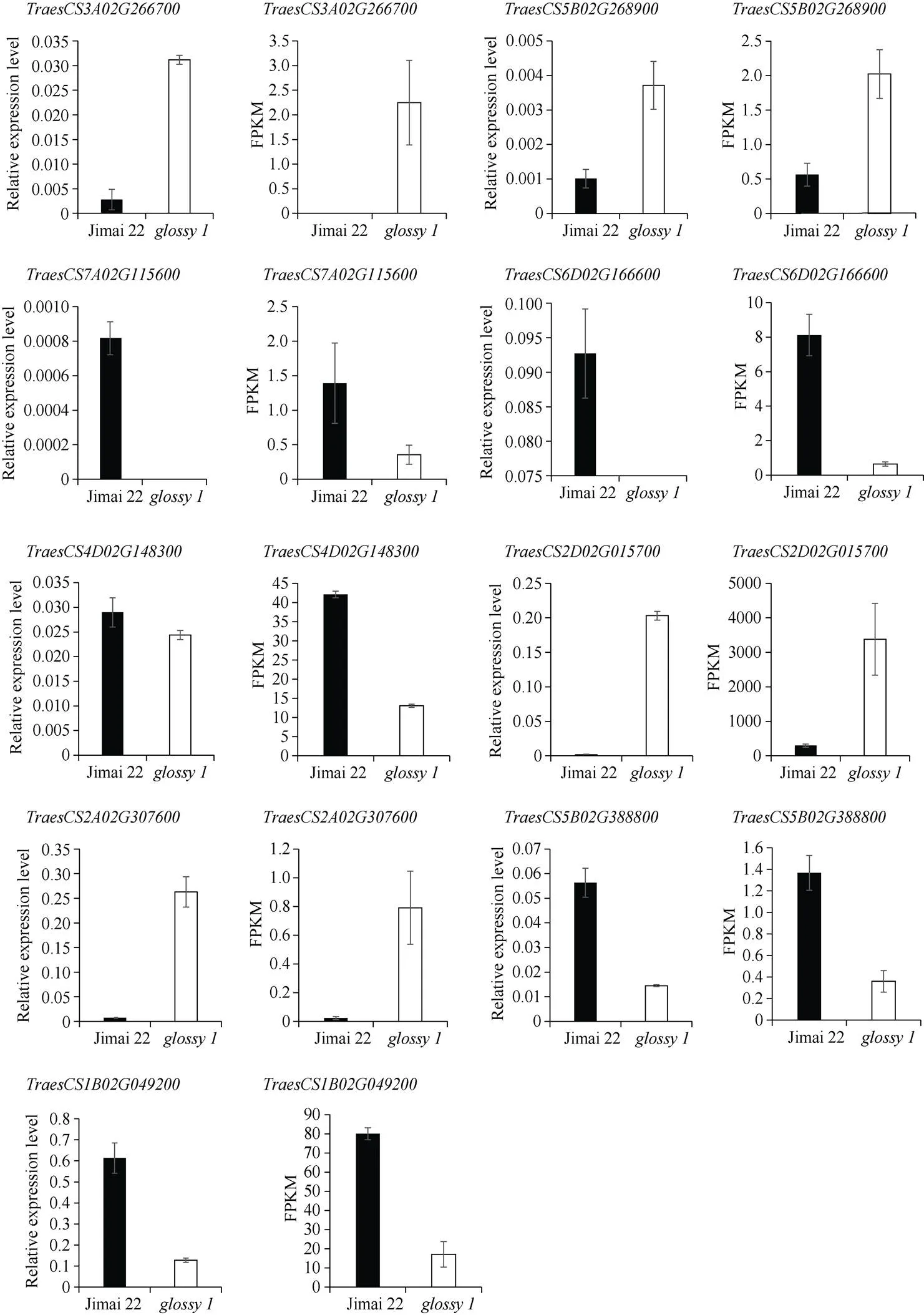
图3 RT-qPCR验证随机挑选的DEGs
为进一步研究小麦颖壳蜡质调控基因的生物学功能, 分别将上调和下调的差异表达基因进行GO富集分析, 根据基因数目分别筛选出30条最显著的条目。结果表明, 上调DEGs主要富集在跨膜转运活性(transmembrane transporter activity)、单加氧酶活性(monooxygenase activity)以及酰基转移酶活性(transferase activity, transferring acyl groups)等条目(图4-B); 下调DEGs中分子功能模块基因数目最多的条目是水解酶活性(hydrolase activity)、过氧化物酶活性(peroxidase activity)以及脂质结合(lipid binding) (图4-C)。
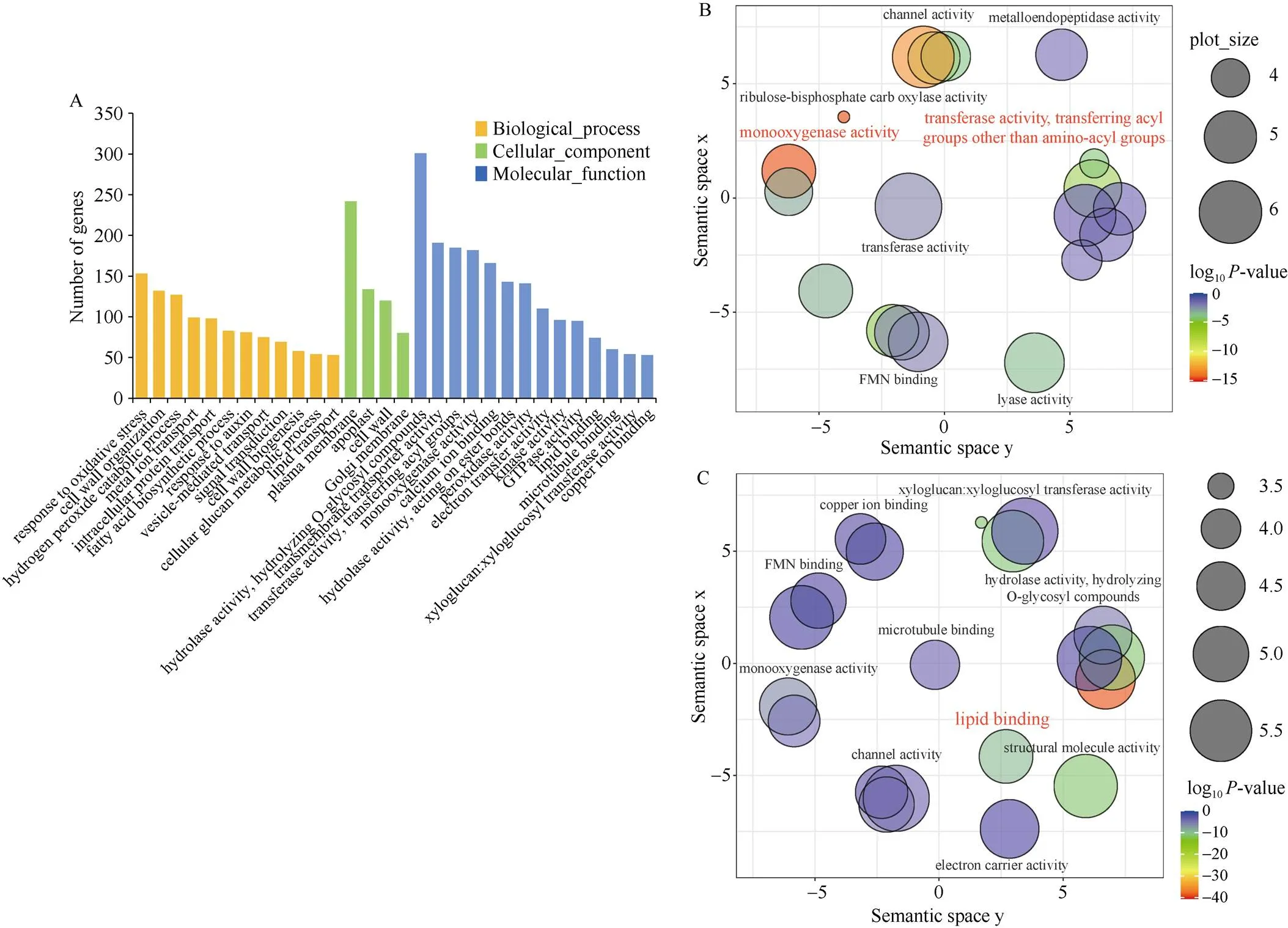
图4 差异表达基因的GO功能注释
A: 差异表达基因的GO功能注释分类; B: 上调DEGs的GO富集; C: 下调DEGs的GO富集。
A: GO functional annotation classification of DEGs; B: GO enrichment of up-regulated DEGs; C: GO enrichment of down-regulated DEGs.
2.5 表皮蜡质合成及转运途径相关基因的表达
表皮蜡质是由多种蜡质化合物组成的, 它的合成需要多步反应。为了从转录组角度分析参与的蜡质代谢途径, 我们结合前人的报道绘制了表皮蜡质合成和转运途径示意图(图5)。从图中我们发现以3-酮酯酰-ACP为底物合成β-二酮途径中涉及的α/β水解酶基因()、查尔酮合酶基因()以及催化羟基β-二酮合成的细胞色素P450基因()在突变体中都上调表达, 这可能导致了突变体中β-二酮含量相对于野生型的显著增加。在合成VLCFAs过程中涉及的长链酰基辅酶A合成酶基因()以及脂肪酸延伸酶复合物中的成员3-酮酰-辅酶A合成酶基因()的表达水平既有上调也有下调, 然而β-羟烷基-辅酶A脱水酶基因()在突变体中上调表达。酰基还原和脱羰途径共有的脂肪酸酰基-辅酶A还原酶基因()在突变体中大部分下调表达, 少部分基因上调表达; 复合物CER1、CER3和CYTB5三者共同作用催化烷烃的合成, 前者涉及的基因在突变体中大部分上调表达, 而后两者均下调表达; 催化初级醇生成蜡酯的O-酰基转移酶家族蛋白基因的表达水平既有上调也有下调。长链脂肪酸及衍生物合成后, 从质膜转运到细胞外间隙过程中涉及的ABC转运G家族蛋白基因()的表达水平在突变体中既有上调也有下调表达, 转运蜡质化合物穿过细胞壁到达角质层外面的脂质转移蛋白基因()在突变体中均下调表达(表3)。这些结果表明在小麦蜡质合成和转运过程中具有非常重要的作用。
为了全面了解调控小麦蜡质代谢的相关途径, 我们根据前人的报道, 进一步在突变体和野生型开花期的颖壳中用RT-qPCR方法检测了五个代谢途径共43个基因的表达情况, 其中25个基因表达水平发生了显著变化, 包括9个脂肪酸延伸途径基因、4个酰基还原途径基因、8个脱羰途径基因、2个转运途径基因以及2个调控途径基因(图6)。结果表明脂肪酸延伸途径、转运及调控途径的基因几乎都下调表达, 而酰基还原及脱羰途径的基因既有上调表达也有下调表达, 这与本实验的转录组结果也是一致的。
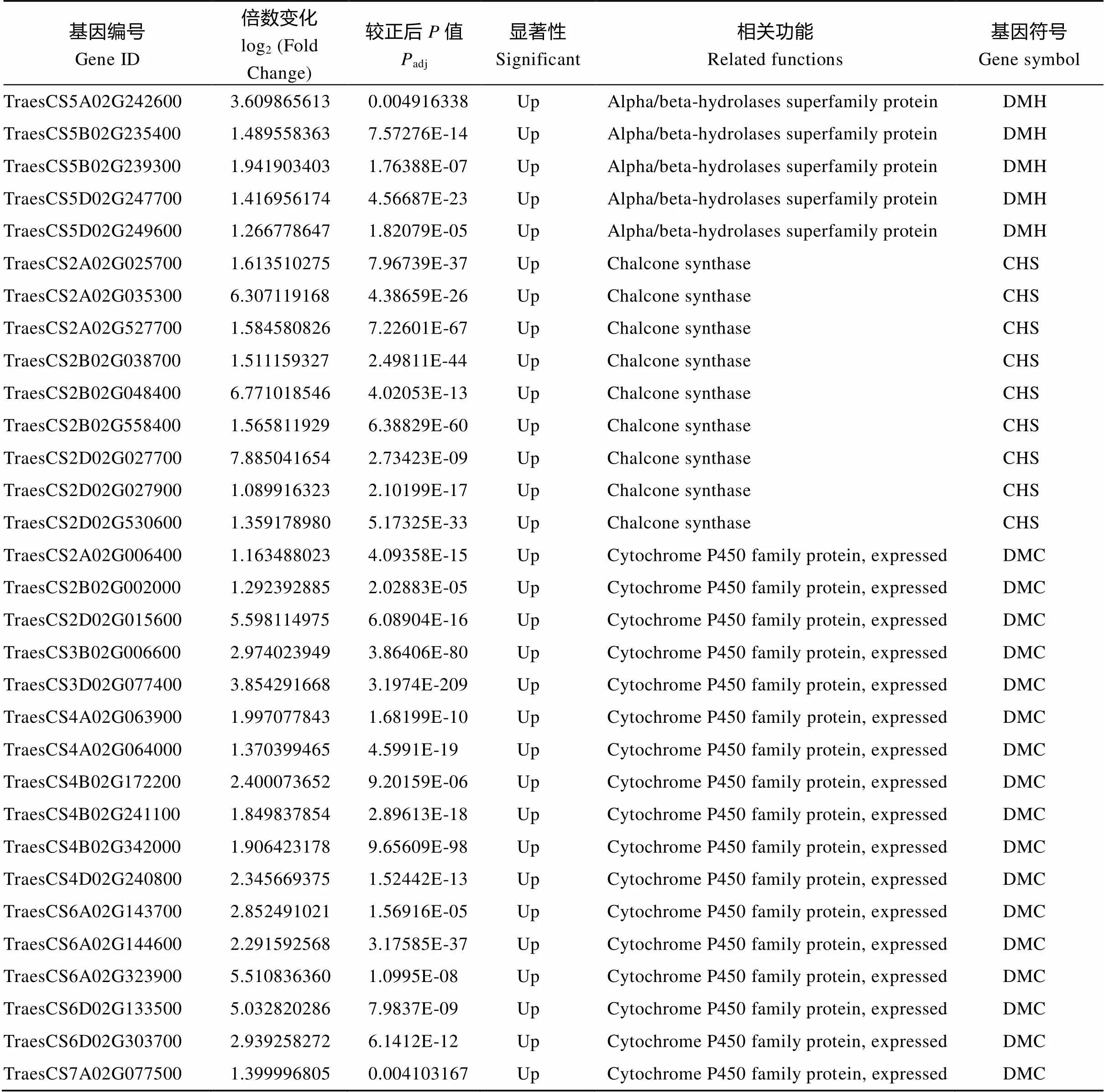
表3 小麦颖壳中蜡生物合成途径相关的差异表达基因
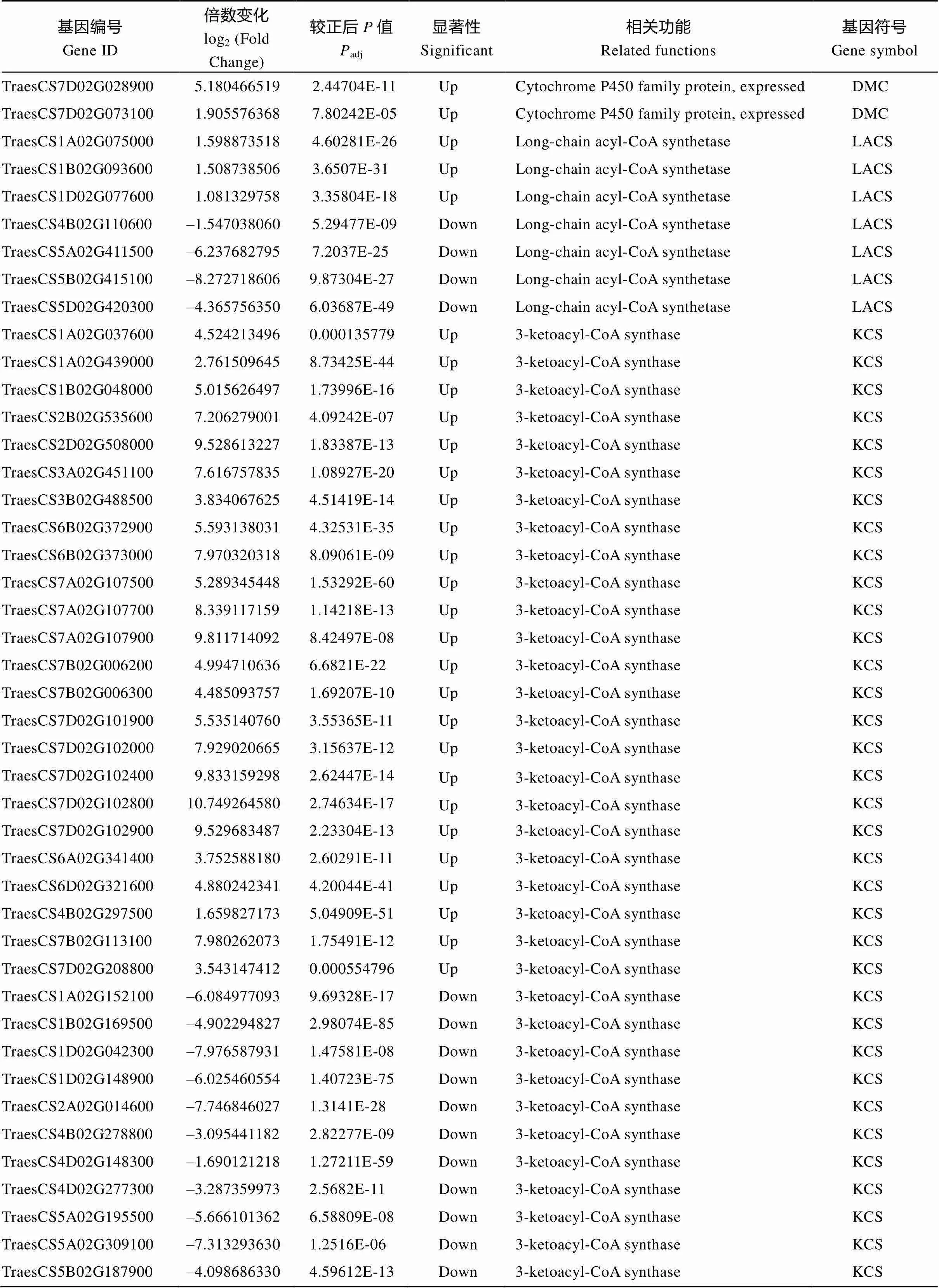
(续表3)
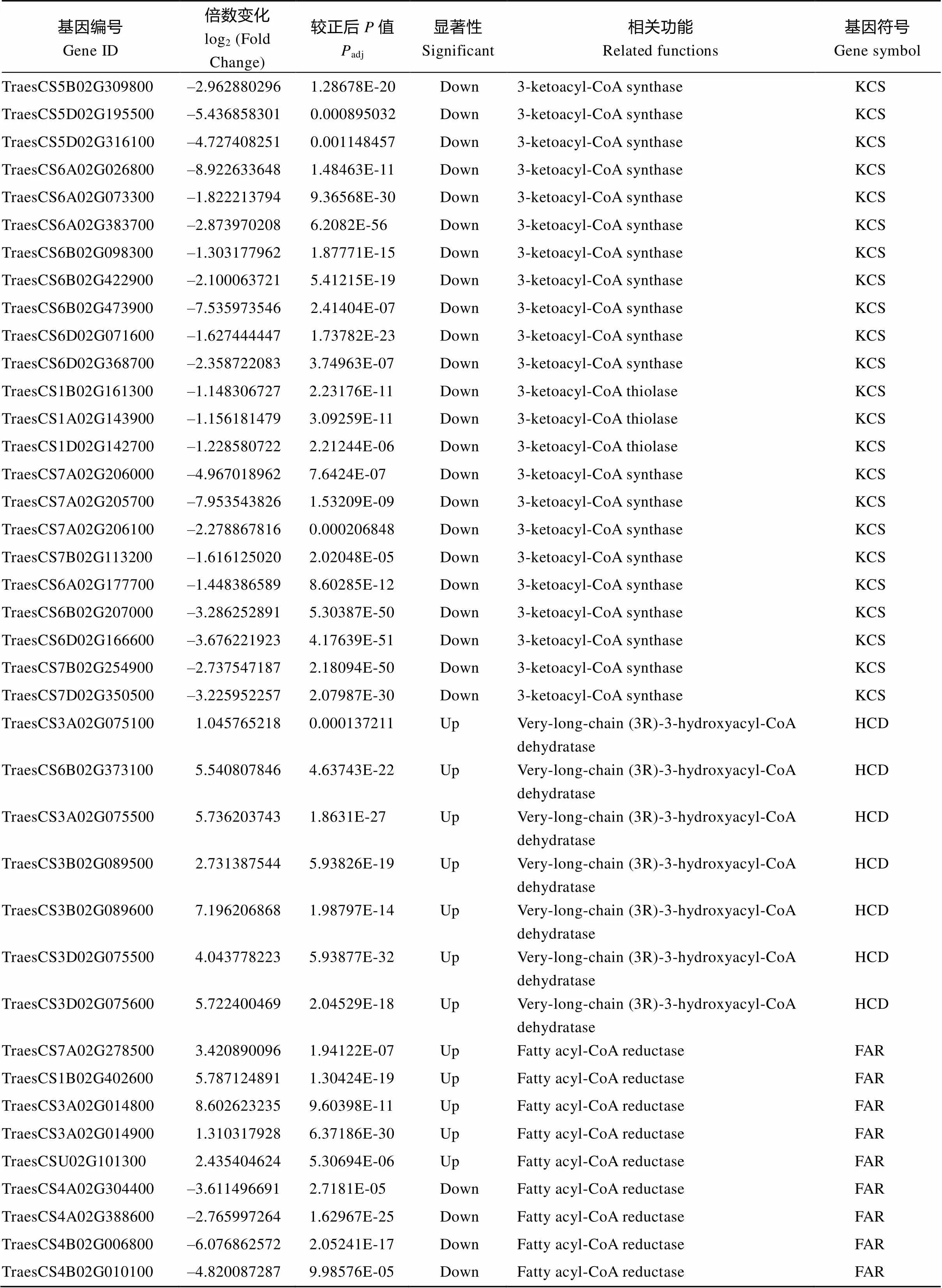
(续表3)
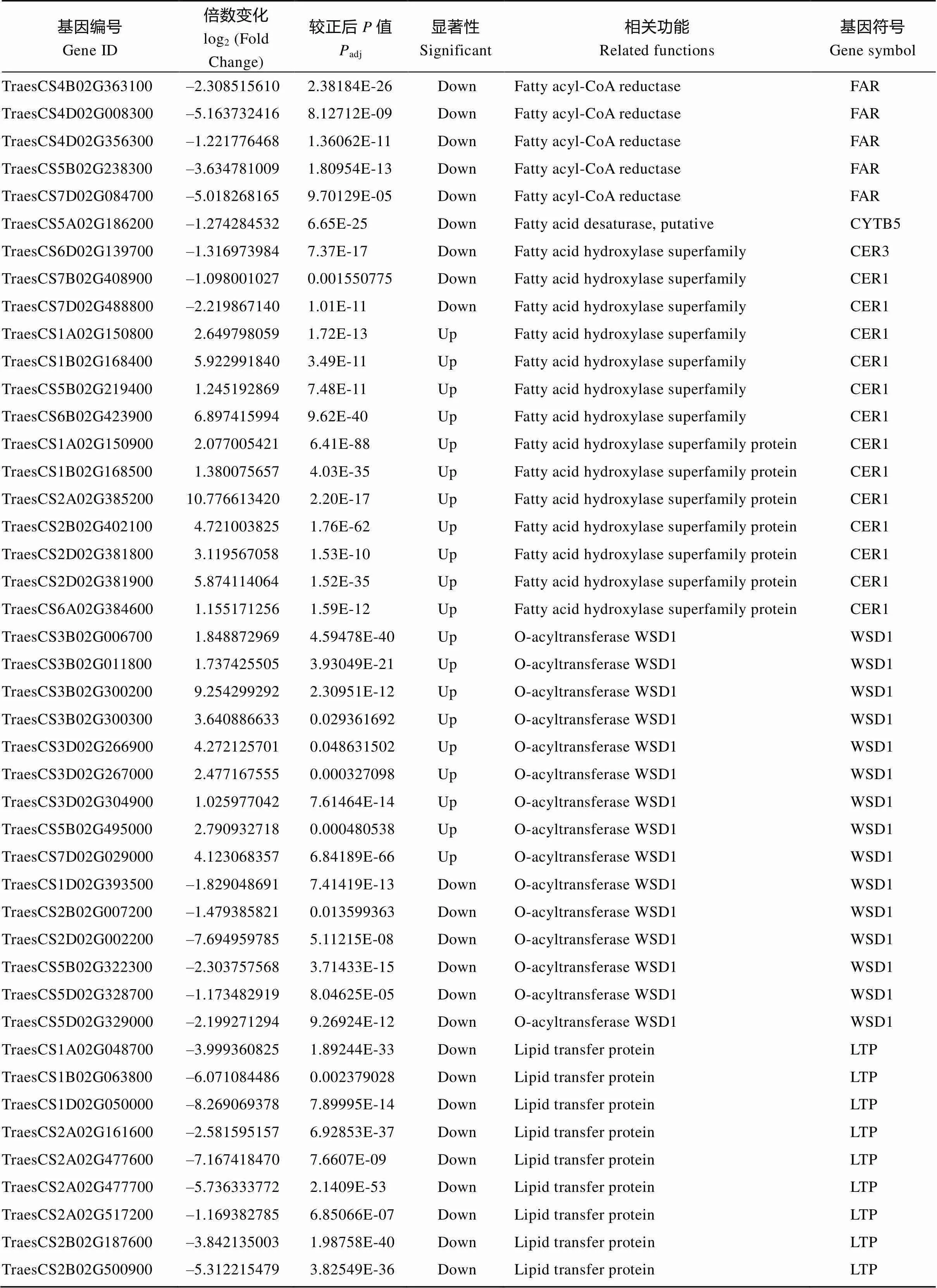
(续表3)
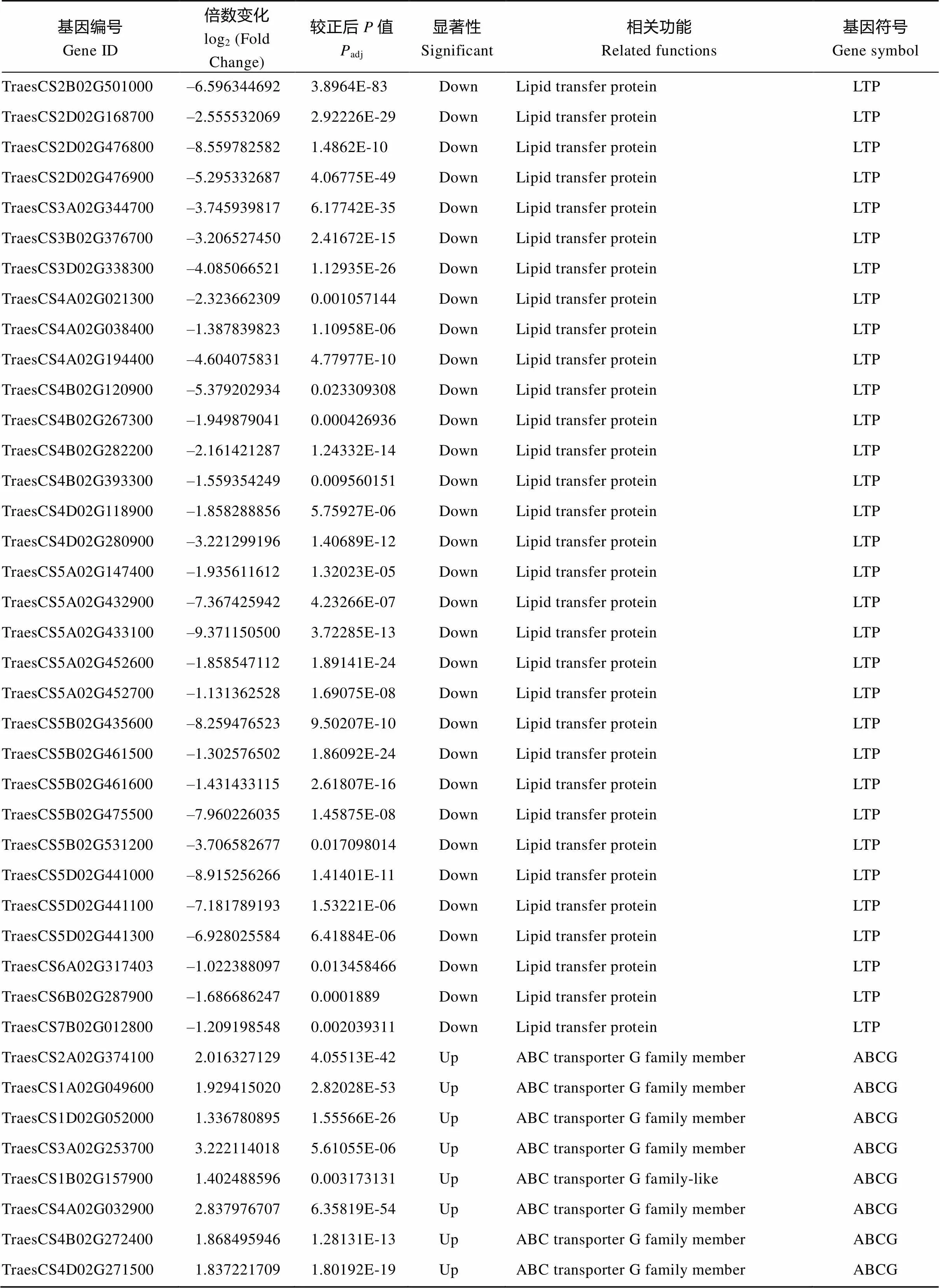
(续表3)
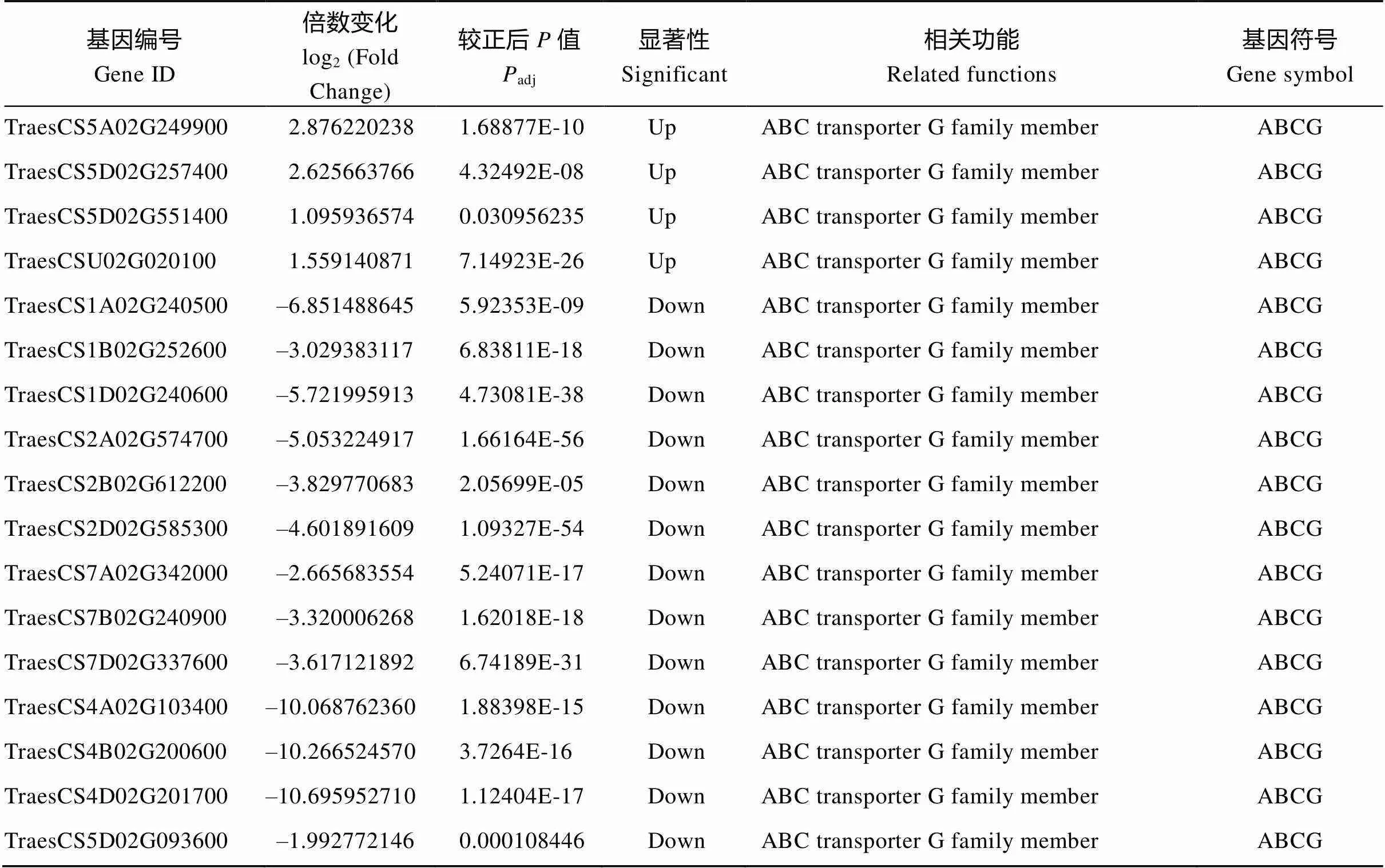
(续表3)
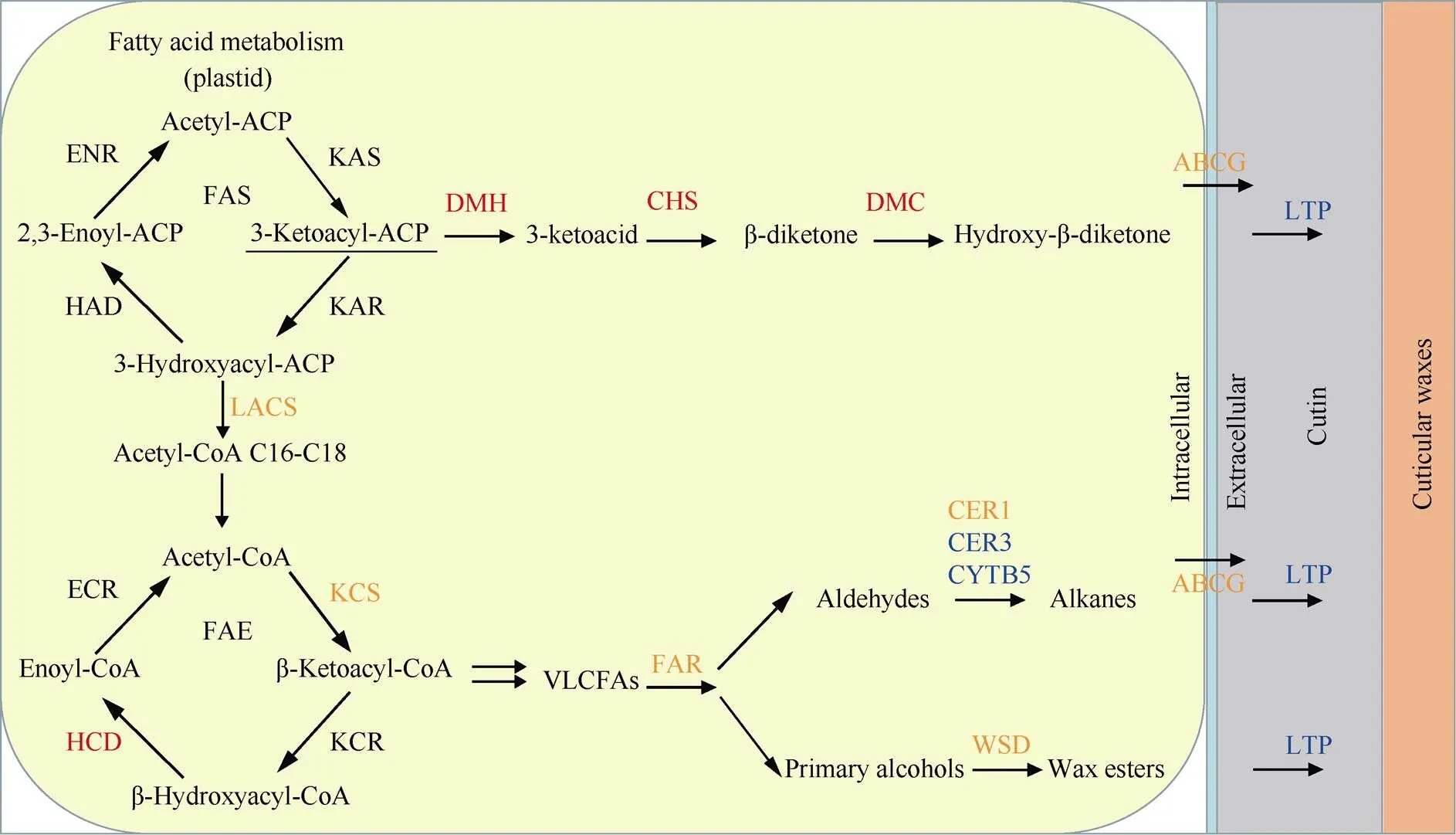
图5 小麦蜡质生物合成示意图
不同颜色的字体表示已知或推测的在表皮蜡质生物合成步骤中起作用的基因表达水平的变化, 其中红色代表突变体相对野生型基因表达上调, 橘色代表这类基因表达水平既有上调也有下调, 蓝色代表下调。
The colored fonts indicate transcriptional changes of the genes known or predicated to be central for epicuticular wax biosynthesis.Compared with the wild-type, the relative expression of the genes in red is up-regulated, the expression of the genes in orange is up-regulated and down-regulated, and the expression of the genes in blue is down-regulated.
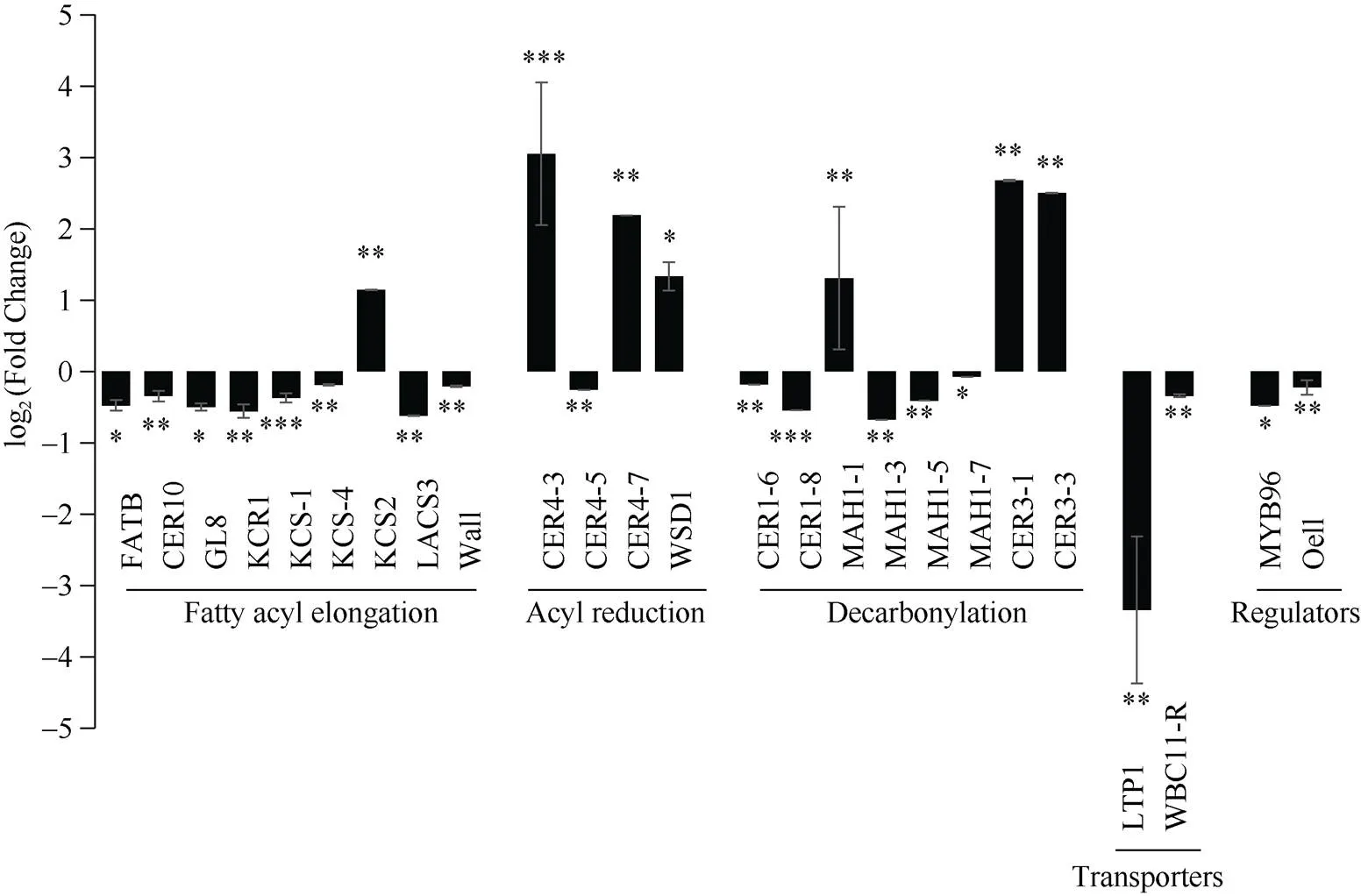
图6 突变体glossy1相对济麦22蜡质基因的转录变化
*:< 0.05; **:< 0.01; ***:< 0.001。
3 讨论
转录组测序(RNA-Seq)技术能够从整体水平研究基因表达差异以及基因结构, 与其他技术手段结合可以解析特定的生物学过程, 已被广泛应用于植物候选基因发掘、功能鉴定及遗传改良等领域。2017年, Huang等利用转录组测序等手段揭示了小麦表皮蜡质合成抑制基因的本质是长链非编码RNA, 并验证了它作为显性抑制因子对蜡质合成基因的调控功能[21]。2021年1月,期刊发文揭示了水稻适应土壤低氮的机制, 研究人员通过多个转录组的交叉比较分析, 从数百个候选基因中发现15个可能受OsTCP19直接调控且与N响应相关的基因, 这为研究的突破性进展指定了正确的方向[22]。
植物表皮蜡质对抵抗外界环境中生物(病原菌、昆虫等)和非生物(干旱、紫外线等)胁迫具有非常重要的作用。目前在小麦中已定位的表皮蜡质基因比较少, 主要包括控制蜡质合成位点~, 抑制蜡质合成位点~, 其中已经克隆的只有两个:和[12,21,23]。因此, 挖掘新的表皮蜡质基因对培育新的小麦抗逆品种是非常重要的。本研究在已定位的颖壳蜡质缺失突变体的基础上开展, 通过对济麦22和突变体开花期的颖壳进行转录组测序, 旨在从转录组水平上揭示突变体中蜡质含量变化的分子机制, 为进一步确定和克隆该基因提供依据, 同时也有助于从全基因组水平上发掘对小麦蜡质代谢有重要作用的功能基因, 为抗逆小麦育种奠定理论基础。
β-二酮是大麦和小麦等小麦族作物表皮蜡质特有的成分, 在拟南芥和其他作物中检测不到, 它的产生决定了普通小麦植株表面呈现白霜状的外观[24-25]。前人已经报道了穗部蜡质合成抑制位点是由于β-二酮的缺失导致颖壳光滑无蜡表型[11]。而本研究通过GO富集分析发现与β-二酮合成相关的基因、和在突变体中均上调表达, 这虽然解释了突变体中颖壳的β-二酮含量相较野生型显著增加的结果, 但是与前人报道的由于β-二酮的缺失导致颖壳光滑无蜡的表型是相悖的。因此深入研究调控机制对于完善小麦蜡质合成代谢机制是很有帮助的。
植物表面白霜状表层是由表皮细胞内质网合成的各种脂质化合物转运到质膜, 再穿过外周细胞壁转运到角质层后自我组装形成的蜡质层。2011年, 陈国雄等克隆了编码ABC转运蛋白G亚家族的一个全转运子HvABCG31的角质层基因, 该基因的功能缺失会导致脂质成分在叶片表皮细胞的大量积累, 导致叶片表面角质层变薄, 角质含量减少[26]。拟南芥中的脂质转运蛋白LTPG参与角质的合成, 该基因突变导致茎表皮细胞角质层蜡质组成发生变化以及蜡质积累减少[14]。此外, 烟草中基因表达量上升, 可使叶片的角质层蜡质积累增加[27]。本研究中ABC转运G家族蛋白基因()和脂质转移蛋白基因()的表达水平在突变体中大部分呈下调表达, 这很可能造成脂质化合物在表皮细胞中积累, 从而导致突变体颖壳蜡质减少。因此, 进一步对蜡质合成及转运途径基因的研究将有助于明确突变体颖壳蜡质缺失的产生原因及潜在的分子机制。
4 结论
本研究利用转录组测序技术系统分析了小麦突变体和野生型颖壳的转录组。通过比较突变体和野生型间的表达水平, 发现了12,230个差异表达基因, 其中5811个基因在突变体中上调表达, 6419个下调表达。通过功能注释分析发现, 这些差异基因主要涉及酰基转移和脂质结合等途径, 这些关键基因的表达水平变化可以解释突变体相较于野生型颖壳蜡质含量的显著变化。本文通过转录组测序的方法揭示了突变体蜡质代谢调控基因的转录图谱, 为深入了解基因功能和小麦蜡质代谢的分子机制及基因调控网络提供了数据信息。
[1] Sturaro M, Motto M, Hemantaranjan A.Plant cuticular waxes: biosynthesis and functions, 2006, 9: 229–251.
[2] Yeats T H, Rose J K C.The formation and function of plant cuticles, 2013, 163: 5–20.
[3] Dehesh K, Tai H, Edwards P, Byrne J, Jaworski J G.Overexpression of 3-ketoacyl-acyl-carrier protein synthase IIIs in plants reduces the rate of lipid synthesis, 2001, 125: 1103–1114.
[4] Leibundgut M, Jenni S, Frick C, Ban N.Structural basis for substrate delivery by acyl carrier protein in the yeast fatty acid synthase, 2007, 316: 288–290.
[5] LiBeisson Y, Shorrosh B, Beisson F, Andersson M X, Arondel V, Bates P D, Baud S, Bird D, DeBono A, Durrett T P.Acyl-lipid metabolism, 2010, 8: e0133.
[6] Bernard A, Domergue F, Pascal S, Jetter R, Renne C, Faure J D, Haslam R P, Napier J A, Lessire R, Joubès J.Reconstitution of plant alkane biosynthesis in yeast demonstrates thatECERIFERUM1 and ECERIFERUM3 are core components of a very-long-chain alkane synthesis complex, 2012, 24: 3106–3118.
[7] Owen R, Huanquan Z, Hepworth S R, Patricia L, Reinhard J, Ljerka K.encodes an alcohol-forming fatty acyl-coenzyme A reductase involved in cuticular wax production in, 2006, 142: 866–877.
[8] Wang M, Wang Y, Wu H, Xu J, Li T, Hegebarth D, Jetter R, Chen L, Wang Z.Three TaFAR genes function in the biosynthesis of primary alcohols and the response to abiotic stresses in, 2016, 6: 25008.
[9] Tulloch A P.Composition of leaf surface waxes ofspecies: variation with age and tissue, 1973, 12: 2225–2232.
[10] Wettstein-Knowles P V, Søgaard B.Theregion in barley: gene cluster or multifunctional gene, 1980, 45: 125–141.
[11] Zhang Z, Wang W, Li W.Genetic interactions underlying the biosynthesis and inhibition of beta-diketones in wheat and their impact on glaucousness and cuticle permeability, 2013, 8: e54129.
[12] Hen-Avivi S, Savin O, Racovita R C, Lee W S, Adamski N M, Malitsky S, Almekias-Siegl E, Levy M, Vautrin S, Berges H, Friedlander G, Kartvelishvily E, Ben-Zvi G, Alkan N, Uauy C, Kanyuka K, Jetter R, Distelfeld A, Aharoni A.A metabolic gene cluster in the wheatand the barleyloci determines beta-diketone biosynthesis and glaucousness, 2016, 28: 1440–1460.
[13] Pighin J A, Huanquan Z, Balakshin L J, Goodman I P, Western T L, Reinhard J, Ljerka K, A Lacey S.Plant cuticular lipid export requires an ABC transporter, 2004, 306: 702–704.
[14] Debono A, Yeats T H, Rose J K C, Bird D, Jetter R, Kunst L, Samuels L.LTPG is a glycosylphosphatidylinositol-anchored lipid transfer protein required for export of lipids to the plant surface, 2009, 21: 1230–1238.
[15] Li L, Chai L, Xu H, Zhai H, Wang T, Zhang M, You M, Peng H, Yao Y, Hu Z, Xin M, Guo W, Sun Q, Chen X, Ni Z.Phenotypic characterization of themutant and fine mapping ofin common wheat (L.), 2021, 134: 835–847.
[16] Marioni J C, Mason C E, Mane S M, Stephens M, Gilad Y.RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays, 2008, 18: 1509–1517.
[17] Love M I, Huber W, Anders S.Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, 2014, 15: 550.
[18] Livak K J, Schmittgen T D.Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCTmethod, 2001, 25: 402–408.
[19] Young M D, Wakefield M J, Smyth G K, Oshlack A.Gene ontology analysis for RNA-seq: accounting for selection bias, 2010, 11: R14.
[20] Chen Y, Song W, Xie X, Wang Z, Guan P, Peng H, Jiao Y, Ni Z, Sun Q, Guo W.A collinearity-incorporating homology inference strategy for connecting emerging assemblies in triticeae tribe as a pilot practice in the plant pangenomic era, 2020, 13: 1694–1708.
[21] Huang D, Feurtado J A, Smith M A, Flatman L K, Koh C, Cutler A J.Long noncoding miRNA gene represses wheat beta- diketone waxes, 2017, 114: E3149–E3158.
[22] Liu Y, Wang H, Jiang Z, Wang W, Xu R, Wang Q, Zhang Z, Li A, Liang Y, Ou S, Liu X, Cao S, Tong H, Wang Y, Zhou F, Liao H, Hu B, Chu C.Genomic basis of geographical adaptation to soil nitrogen in rice, 2021, 590: 600–605.
[23] Li L, Qi Z, Chai L, Chen Z, Wang T, Zhang M, You M, Peng H, Yao Y, Hu Z, Xin M, Guo W, Sun Q, Ni Z.The semidominant mutationimpairs epicuticular wax deposition in common wheat (L.), 2020, 133: 1213–1225.
[24] Adamski N M, Bush M S, Simmonds J, Turner A S, Mugford S G, Jones A, Findlay K, Pedentchouk N, von Wettstein-Knowles P, Uauy C.The inhibitor oflocus () prevents formation of beta- and OH-beta-diketones in wheat cuticular waxes and maps to a sub-cM interval on chromosome arm 2BS, 2013, 74: 989–1002.
[25] Bianchi G, Figini M L.Epicuticular waxes of glaucous and nonglaucous durum wheat lines, 1986, 34: 429–433.
[26] Chen G, Komatsuda T, Ma J F, Nawrath C, Pourkheirandish M, Tagiri A, Hu Y G, Sameri M, Li X, Zhao X, Liu Y, Li C, Ma X, Wang A, Nair S, Wang N, Miyao A, Sakuma S, Yamaji N, Zheng X, Nevo E.An ATP-binding cassette subfamily G full transporter is essential for the retention of leaf water in both wild barley and rice, 2011, 108: 12354–12359.
[27] Cameron K D, Teece M A, Smart L B.Increased accumulation of cuticular wax and expression of lipid transfer protein in response to periodic drying events in leaves of tree tobacco, 2006, 140: 176–183.
Transcriptome profiling ofmutant with glossy glume in common wheat (L.)
LI Ling-Hong, ZHANG Zhe, CHEN Yong-Ming, YOU Ming-Shan, NI Zhong-Fu, and XING Jie-Wen*
College of Agronomy, China Agricultural University, Beijing 100193, China
In order to clarify the molecular mechanism of the significant change in glume wax content, transcriptome profiling was performed on glossy glume mutantand its wild-type Jimai 22.The results showed that a total of 12,230 differentially expressed genes were screened in themutant, among which 5811 genes were up-regulated and 6419 were down-regulated.GO functional enrichment revealed that the differentially expressed genes were mainly enriched in the wax synthesis and transport pathway, specifically distributed in acyltransferase activity, lipid binding, hydrolase activity.The findings suggested that these pathways were closely related to the glume-wax-deficient trait in wheat.We also detected the relative expression levels of some genes involved in the wax metabolic pathway by RT-qPCR, and the results were consistent with the transcriptome data.This study not only provides data support for the further study of the molecular mechanism of wax metabolism and gene regulation network but also lays a theoretical foundation for the breeding of stress-resistant variety in wheat.
wheat; epicuticular wax;mutant; transcriptome analysis; molecular mechanism
2021-01-12;
2021-04-14;
2021-05-21.
10.3724/SP.J.1006.2022.11006
通信作者(Corresponding author): 邢界文, E-mail: Jiewen.Xing@cau.edu.cn
E-mail: lilinghong00en@163.com
本研究由国家自然科学基金项目(31991214)和国家重点研发计划项目(2017YFD0101004)资助。
This study was supported by the National Natural Science Foundation of China (31991214) and the National Key Research and Development Program of China (2017YFD0101004).
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20210521.1245.002.html
