锁核酸和不对称扩增辅助的等位基因多重引物识别KRAS基因突变的研究
2022-11-04刘义庆褚福禄兰文军
师 声,张 静,胡 岳,刘义庆,褚福禄,兰文军*
1.齐鲁工业大学(山东省科学院) 生物工程学院,山东 济南 250353;2.山东第一医科大学附属省立医院临床医学检验部,山东 济南 250021
结直肠癌是一种起源于结肠或直肠的胃肠道恶性肿瘤,是世界上发病率第三高的恶性肿瘤,(185万新发病例/年;占恶性肿瘤总数的10.2%)。人口老龄化、高收入导致的饮食习惯变化以及缺乏体育锻炼、过度肥胖和不良嗜好等因素导致结直肠癌发病率逐年上升。目前0~74岁人群的累积发病风险为2.27%,而年龄在50岁至85岁之间患病率增加了10倍以上。与女性相比,男性的风险高出了50%以上(男性女性0~74岁的风险分别为2.75%和1.83%)[1]。大约35%的患者在诊断时表现为转移性疾病,多达50%的非转移性结直肠癌患者最终表现为转移性疾病[2]。由于伴随诊断技术的应用,转移性CRC(mCRC)的个性化治疗已经取得了显著进展。
大鼠肉瘤病毒致癌基因同源物(RAS)中的单核苷酸变异被分类为Kirsten RAS(KRAS)、Neuroblastoma RAS(NRAS)和Harvey RAS(HRAS),已被确定为指导转移性结直肠癌治疗药物使用的生物标志物。癌基因RAS编码一种三磷酸鸟苷(GTP)结合蛋白(GTPase),并在表皮生长因子受体(EGFR)触发的RAS-RAF-MEK-ERK信号级联中占据重要地位。驱动肿瘤生长,致癌转移和免疫抑制[3-4]。KRAS[5]、NRAS[6]和HRAS[7]突变率分别为40%、4%和<1%。此外,KRAS突变也与姑息性原发性肿瘤切除术(PTR)手术中不可切除的mCRC患者的生存结果相关[8]。
利用碱基错配、并在突变鉴别反应孔和参考反应孔中使用共水解探针和反向引物,基于单重引物等位基因鉴别分析[9]以及多重引物等位基因鉴别分析[10]的KRAS基因突变检测已有报道。然而,我们发现上述多重分析中的ΔCq值(突变检测与参考检测阈值周期的差异)并不能正确关联结果。在锁定核酸(LNA)和不对称扩增的辅助下,本研究设计、优化和评价了一种检测KRAS突变的多重引物等位基因识别(MPmAD)分析,可用于指导mCRC治疗中anti-EGFR单克隆抗体的给药。
1 实验仪器与材料
1.1 实验仪器
ABI 7500 荧光定量PCR仪(美国applied biosystems公司);5804R高速冷冻离心机(德国Eppendorf公司);IMPLEN 5186超微量分光光度计(德国Implen公司);epMotion 5075自动移液工作站(德国Eppendorf公司);DYY-6C 核酸电泳仪(北京六一生物科技有限公司);EVOS M7000全自动活细胞成像系统(美国Thermofisher公司);NuGenius 凝胶成像系统(英国syngene公司)。
1.2 实验材料
多重前向引物、参考前向引物、反向引物和探针自行设计,并由生物工程(上海)股份有限公司合成。所有引物和探针序列如表1、表2所示。

表1 多重检测体系引物/探针序列

表2 参考体系引物/探针序列
使用外周血基因组DNA提取试剂盒(天根,DP304-02)提取野生型基因组DNA。本研究福尔马林固定石蜡包埋组织样本取自山东大学齐鲁医院、山东第一医科大学。使用QIAamp DNA石蜡组织试剂盒(No.1080391)提取福尔马林固定石蜡包埋组织DNA。超微量分光光度计检测提取DNA浓度,提取DNA于-20 ℃保存。
2 方法与结果
2.1 方法
2.1.1 KRAS突变检测设计策略
为检测KRAS突变(G12R、G12S、G12C、G12A、G12D、G12V和G13D),多重检测体系中使用了等位基因特异性(AS)正向引物、FAM-BHQ1标记的水解探针和反向引物,参考体系中使用了参考正向引物、FAM-BHQ1标记的探针和反向引物,设计策略如图1所示。通过计算ΔCq值(多重检测试验和参考试验之间阈值循环数的差异),以确定阴性和阳性。由于锁核酸(LNA)引物在多重检测体系中表现出更高的识别能力和特异性[11],我们在引物3′端利用LNA修饰了多重前向引物。在本研究中,内参被省略,参考分析中的扩增子起到模板质量控制的作用。
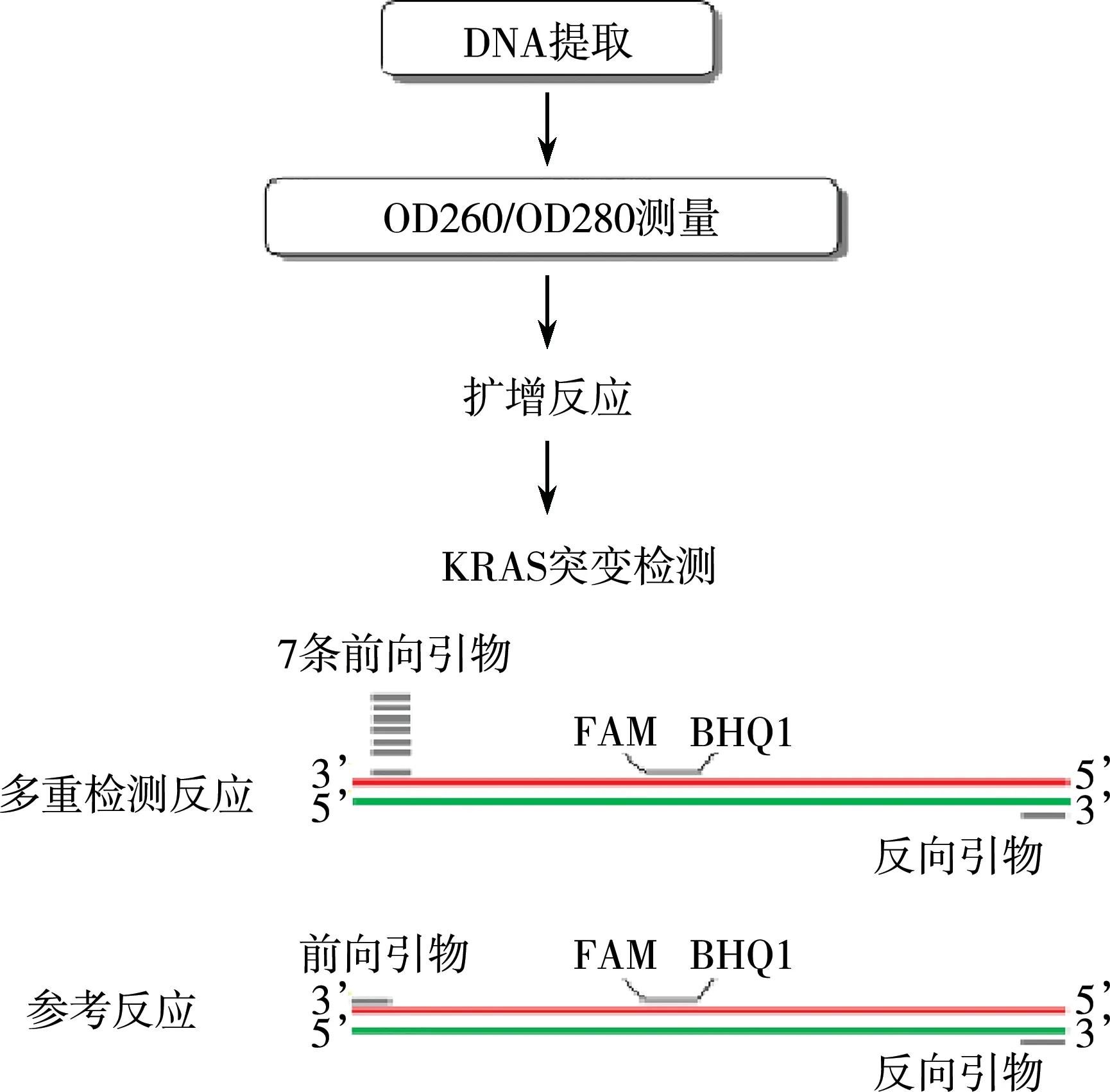
图1 KRAS突变的MPmAD分析策略
2.1.2 核酸实时扩增
想要避免一味比较的习惯,真正有效的办法不是禁止做某事,而是用做另一件事来替代。如果我们要减少有害的比较,就得让自己去做有益的比较。
多重检测扩增反应体系为20 μL,包含10 μL 2×TaqMan master mix,0.4 μmol/L锁核酸修饰的前向引物(F PCR primer-G12R-G13D)、0.4 μmol/L反向引物(R PCR primer),0.2 μmol/L水解探针(PCR probe)和50 ng DNA模板。参考扩增反应体系则使用0.4 μmol/L参考前向引物(F-reference PCR primer)代替锁核酸修饰的前向引物。使用ABI 7500仪器进行反应,采用两步法,95 ℃预变性1 min,95 ℃变性15 s,60 ℃退火/延伸30 s,反应进行40循环。每组实验至少包括一个阳性对照和一个不含DNA的阴性对照。
2.1.3 预估阈值
重复测量10个野生型基因组DNA样本,获取多重反应与参考反应扩增循环数差值ΔCq,ΔCq检测阈值=ΔCq最小值-1。当ΔCq检测阈值<11时,KRAS检测为阳性。
2.1.4 一致性分析
FFPET标本送至青岛派森诺基因生物技术有限公司进行Sanger测序,使用Kappa系数检验本方法与Sanger测序两种方法学的一致性。
2.2 结果
2.2.1 灵敏度
以突变型质粒分子模型对检测体系进行灵敏度分析。在50 ng野生型基因组DNA背景下,制备KRAS 12/13密码子10.0%、3.0%、1.0%和0.3%突变型质粒分子模型,并用MPmAD检测方法分析质粒混合样本。为优化分析灵敏度,多重分析采用不对称扩增法,将F-primer G12C、G12D和G12V的浓度分别调整为1.0、1.2和1.0 μmol/L。如图2显示,KRAS突变的检测限(LOD)至少为3%。
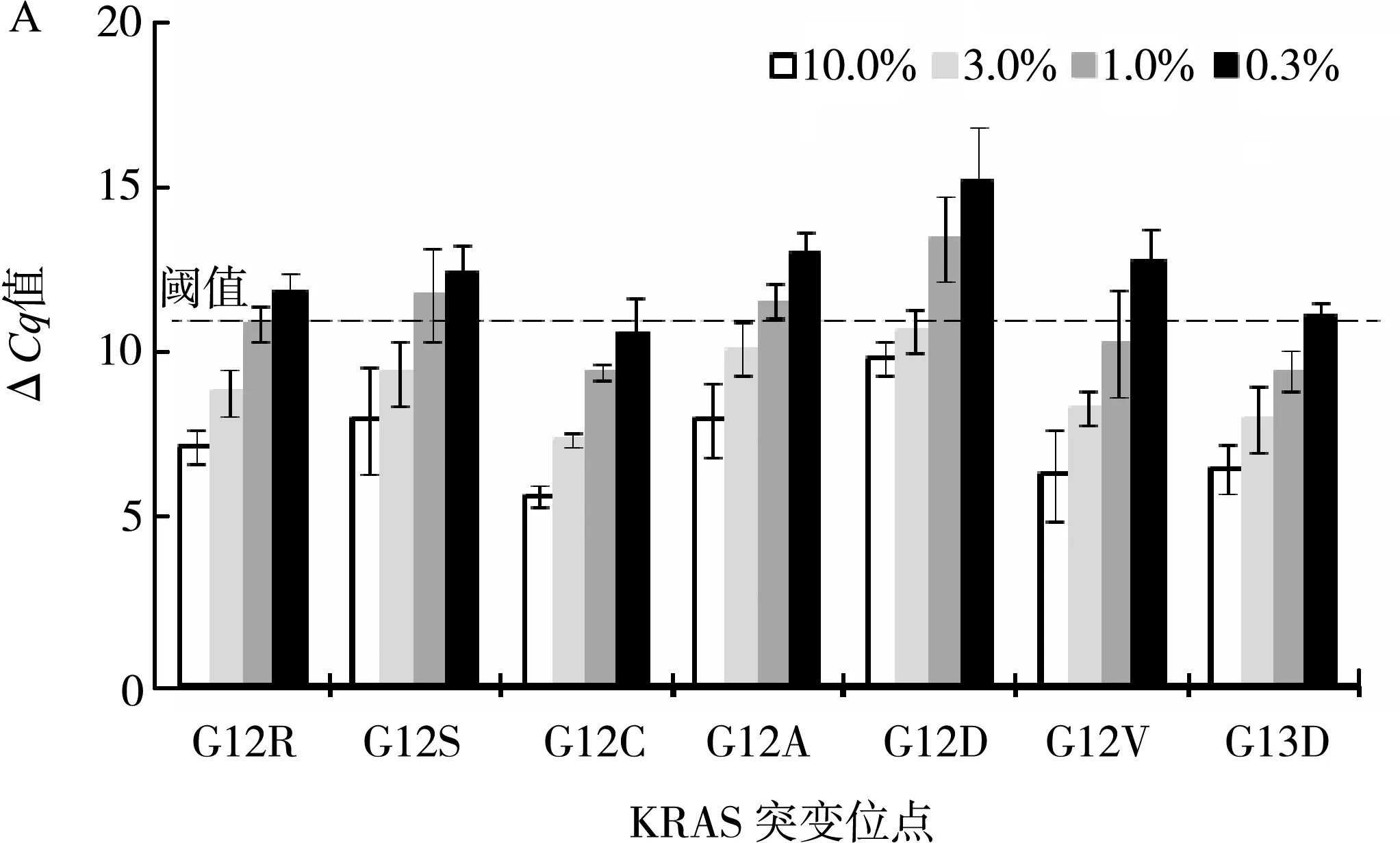
图2 MPmAD分析灵敏度
按照多重检测方法进行普通PCR反应。将50 ng野生型基因组DNA背景的10% KRAS突变质粒的扩增子在1.5%琼脂糖凝胶上电泳,验证其特异性。电泳结果显示,质粒混合物有扩增条带,而野生型基因组DNA没有扩增条带,如图3A所示。
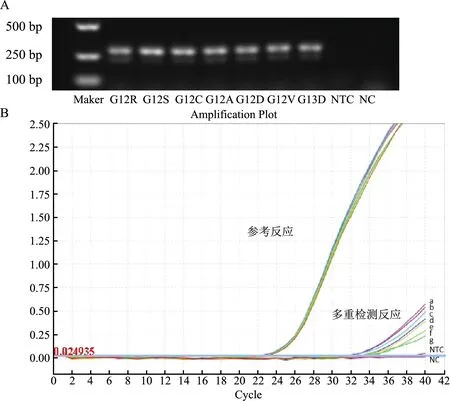
A—10%的KRAS突变型质粒分子模型扩增子的1.5%琼脂糖凝胶电泳图(NTC:阴性野生型基因组DNA;NC:无DNA阴性对照);B— 3%的KRAS突变型质粒多重检测反应和参考反应扩增图(a,b,c,d,e,f,g:G13D,G12A,G12V,G12C,G12R,G12S和G12D)。图3 MPmAD分析灵敏度
图3B显示,与有野生型基因组DNA(negative control with wild-type genomic DNA,NTC)或没有野生型基因组DNA的阴性对照(negative control without DNA,NC)相比,3%突变KRAS12/13密码子的混合质粒样本显著扩增。
2.2.3 精密度
通过检测3%突变的KRAS 12/13密码子质粒来评估检测试剂精密度。此外,我们还检测了2个携带KRAS 12/13密码子上突变的FFPET样本。2人操作5天内每天使用两批次进行测试,每个标本每批次进行8个重复测试(n=80/标本),共收集80个ΔCq值,计算变异系数(CV)=标准差/平均值×100%。精密度评估显示,3%G12R、G12S、G12C、G12A、G12D、G12V、G13DKRAS质粒样本CV值分别为:7.74%、5.89%、8.20%、7.95%、7.33%、8.16%、7.39%;NO.42 FFPET、NO.5 FFPET样本变异系数分别为:10.94%、6.79%。所有天数、样本、重复品、操作人员和试剂批次组合的CV值均<15%。
2.2.4 交叉实验
本研究通过判断同源质粒的存在是否影响KRAS突变检测来评价交叉反应。对野生型基因组DNA样本和野生型基因组DNA背景的3%突变质粒样本,分别混合3%的KRAS同源(KRAS12/13密码子假基因、NRAS2号外显子及HRAS2号外显子)质粒。结果表明,同源质粒在本实验中无交叉反应。
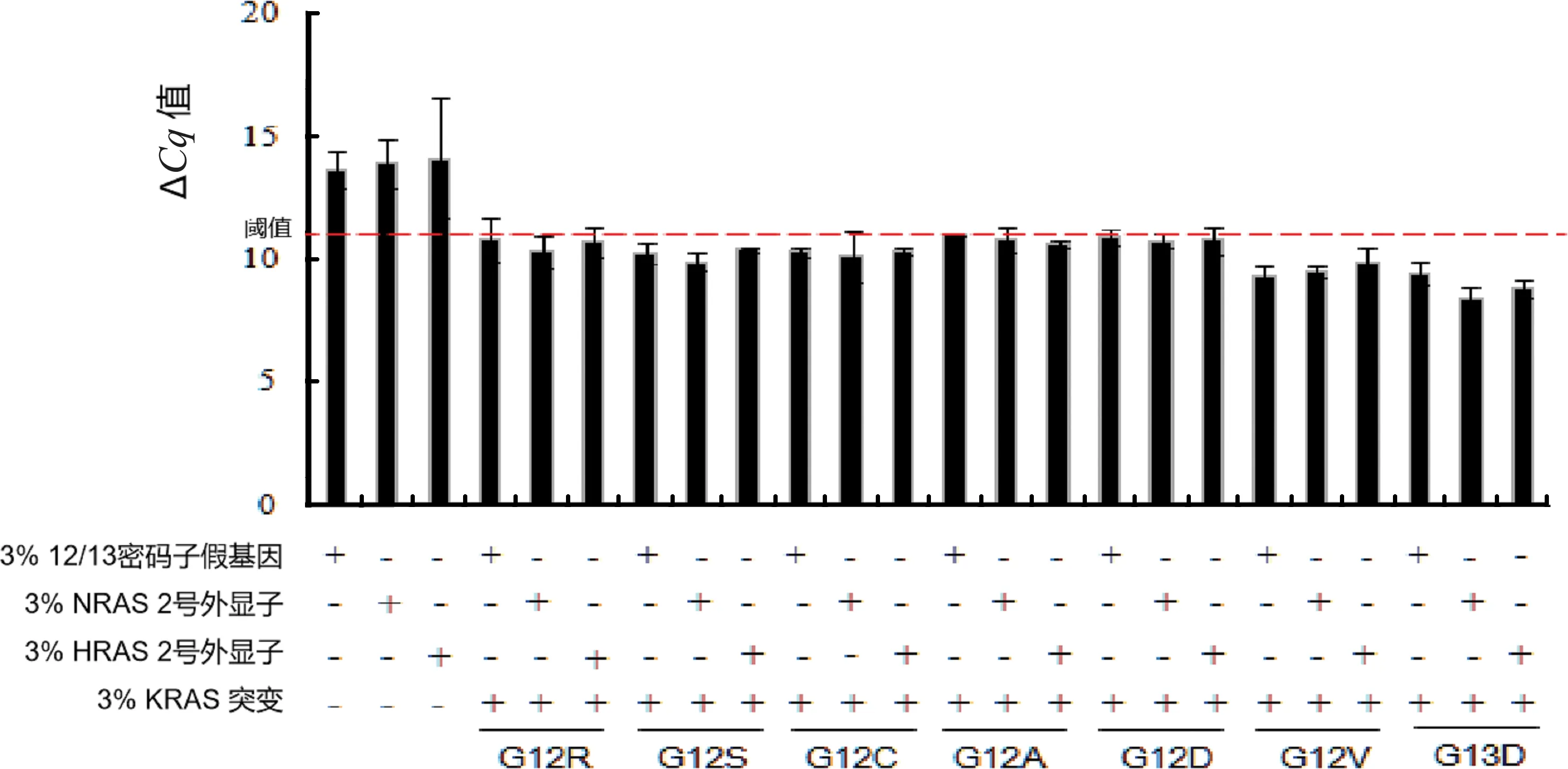
图4 MPmAD分析交叉实验结果
2.2.5 一致性分析
本检测方法与Sanger测序对KRAS突变的鉴别无统计学差异(k=0.946;p<0.001)。在包含115个FFPET样本中,共有70个样本被本检测方式检测为阳性。其中有3例阳性样本被本方法检出,而Sanger测序显示为阴性。以Sanger测序为参考,计算出的阳性一致性为100.00%(95%CI 93.51%~100%),阴性一致性为93.75%(95%CI 82.53%~98.49%),一致性结果如表3,图5A、B所示。
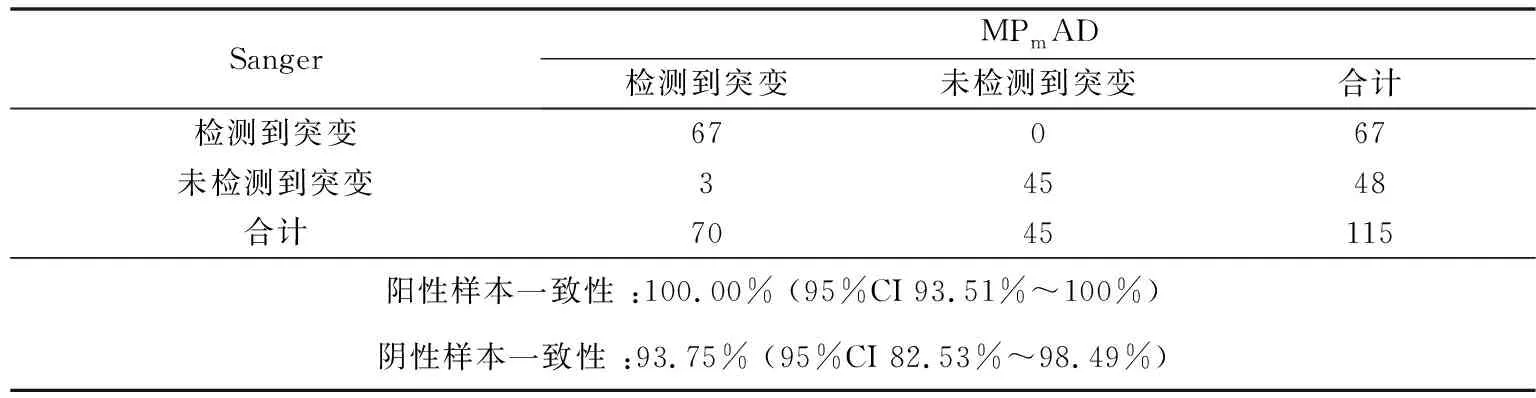
表 MPmAD与Sanger测序对KRAS基因检测的一致性分析
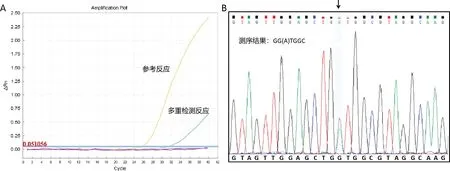
图5 MPmAD代表扩增与Sanger测序图
3 讨 论
在人类DNA单核苷酸多态性靶点的多重AS-PCR中,LNA引物具有更高的特异性。本研究的AS引物在多重检测体系中中用LNA修饰。LNA是一种带有2′-O,4′-C-亚甲基的核酸衍生物[12]。生成的双环结构将核糖部分锁定为C3′-endo构象,这可以增强杂化Tm[13]和杂交特异性[14]。特异性的提高可能是由于具有3′LNA末端的AS引物形成了较少的模板连接,并影响了DNA聚合酶底物的利用[15]。
由于从FFPET中提取的DNA容易降解为100~200 bp的片段,本研究使用ΔCq值来决定结果,以避免假阴性。在这项研究中,不对称扩增也用于提高G12C、G12D、G12V和Q61R的分析灵敏度。结果显示,KRAS检测灵敏度至少为3%。检测115例FFPET样本,有3例阳性样本可被本方法检出,而Sanger测序显示为“阴性”,这可能是由于Sanger测序灵敏度(10%)较低所致。本研究表明,与Sanger测序与融解曲线分析法(cobas©KRAS Mutation Test,Roche Diagnostics)相比,锁核酸和不对称扩增辅助的多重引物等位基因识别分析更敏感。
由于在实时荧光PCR中共扩增时,目标反应可能强于内控反应。本研究省略了内参对照品,将参考反应扩增子用于ΔCq值的获取和模板的质控。
伴随诊断试剂盒的非临床评价的分子模型主要包括:(i)基因组DNA背景下的质粒混合;(ⅱ)细胞系/FFPET的突变和野生型DNA的混合物。由于突变的细胞系和FFPET标本不易收集,本研究采用了质粒混合物[16]评估分析的灵敏度、交叉反应性和重现性。
据COSMIC数据库,12/13密码子上的7个KRAS热点突变占所有KRAS突变的92%以上。研究证明,7个KRAS热点突变可以清楚地预测西妥昔单抗联合FOLFOX/folfiri治疗的反应[17-18]。这些数据表明,检测7个KRAS热点突变足以在mCRC中指导anti-EGFR单克隆抗体的使用。
本研究开发了一种基于锁核酸和不对称扩增辅助多重引物等位基因分析方法,可用于指导anti-EGFR单克隆抗体在mCRC治疗中的靶向给药,该方法具有简洁和低成本的优点。与多探针等位基因鉴别分析相比[19],本研究中描述的检测方法更加经济,尤其适用突变体比例可能很低的FFPET样本[20]。
