Rosai-Dorfman病12例临床病理及分子特征
2022-11-04王清扬王晓伟钟定荣
王清扬,王晓伟,钟定荣
Rosai-Dorfman病(Rosai-Dorfman disease, RDD)又称为伴巨大淋巴结病的窦性组织细胞增多症,是一种罕见的、原因不明的非朗格汉斯细胞组织细胞增多症。RDD曾被认为是一种反应性、非肿瘤性疾病,近年来部分支持RDD克隆性的证据已被发现,但由于RDD较为罕见,伴有基因改变的RDD常为个案报道。二代测序技术(next-generation sequencing, NGS)实现了全基因组高通量、低成本和快速化检测,已在生物医学领域广泛应用。因此,本文回顾性分析12例RDD的临床病理资料及随访材料,行免疫组化染色及分子检测,并结合文献复习以提高对该病的认识。
1 材料与方法
1.1 临床资料收集中日友好医院病理科2012年6月~2020年12月会诊及收治的RDD共12例,对其临床病理资料进行分析,并通过门诊复查、病案查询及电话形式进行随访。
1.2 方法
1.2.1免疫组化 将手术切除及穿刺活检标本经10%福尔马林固定,常规脱水、石蜡包埋、切片,HE染色,镜下观察。免疫组化染色采用EnVision法,一抗包括S-100、CD68、CD138、CD38、CD3、CD5、CD20、Ki-67、CD1a、Cyclin D1(所用试剂盒购自罗氏公司),选用组织坏死性淋巴结炎2例及朗格汉斯组织细胞增生症(Langerhans cell histiocytosis, LCH)2例作为对照。
1.2.2分子检测 采用基于杂交捕获的高通量测序法(Hybridization Capture-based NGS)。测序流程包括,(1)DNA提取:石蜡包埋组织10 μm厚连续切片8张,放入1.5 mL离心管,二甲苯脱蜡后使用艾德生物公司FFPE DNA试剂盒按照说明书进行标本DNA提取,溶解于30~100 μL DTE缓冲液中,取适量样品,对DNA质量浓度进行测定。(2)文库构建:片段化处理、末端修复、接头连接、PCR扩增、杂交捕获。使用Illumina Nextseq 500进行基因检测,采用艾德生物公司人类癌症多基因突变联合检测数据分析系统进行分析,覆盖116个基因的外显子、剪切区域、融合相关内含子。判断阳性条件:平均有效测序深度>500×,基因突变比例≥0.4%,突变绝对拷贝数≥2,链平衡性0.1~10。
2 结果
2.1 临床特征12例RDD中,男性8例,女性4例,男女比为2∶1,平均年龄40岁(8~80岁)。2例淋巴结型,9例结外型(中枢神经系统2例、皮肤2例、鼻部2例、气管1例、上纵隔1例、乳腺1例),1例混合型(累及下颌、腹部皮肤及腋下淋巴结)。12例患者均接受手术治疗,7例术后接受激素冲击治疗。9例患者获得随访资料,中位随访时间37个月(7~109个月),4例患者术后6~36个月复发,1例活检病例缓解,3例无复发,1例不典型病例4年后死亡(表1)。

表1 12例RDD的临床资料
2.2 病理检查眼观:12例中2例为穿刺活检标本,1例送检灰白色破碎组织,余肿物均呈结节状,无包膜,大小1.5 cm×0.8 cm×0.1 cm~4.0 cm×3.0 cm×2.0 cm,切面灰白或灰黄色,实性、质中。镜检:低倍镜下淋巴结型RDD淋巴结结构破坏,皮肤RDD病变位于真皮中,淋巴结外病变常伴有纤维组织增生。中高倍镜下病变部位可见大量浆细胞、较多淋巴细胞及组织细胞一起呈现出交替的“暗区”和“亮区”(图1)。“亮区”由病变组织细胞构成,细胞核呈圆形或椭圆形,胞质丰富、嗜酸性,部分组织细胞胞质内吞噬淋巴细胞、浆细胞(即“伸入现象”)(图2),其中1例可见吞噬红细胞和炎细胞的现象;“暗区”主要为增生成片的浆细胞和淋巴细胞构成。淋巴结内病变吞噬现象比结外病变常见。
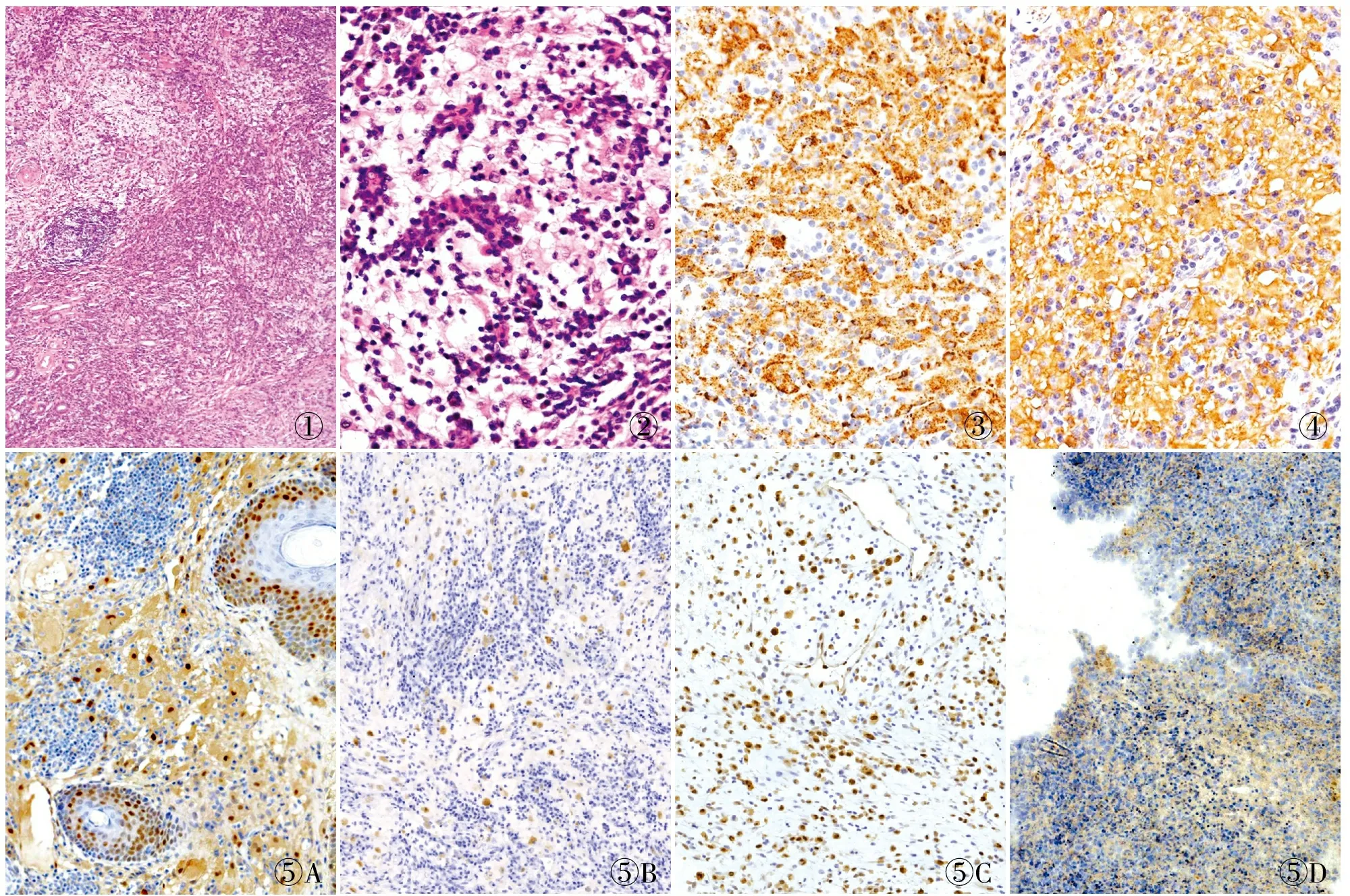
图1 慢性炎细胞及组织细胞呈现出交替的“暗区”和“亮区” 图2 组织细胞胞质内可见淋巴细胞、浆细胞“伸入现象” 图3 病变组织细胞CD68阳性,EnVision法 图4 病变组织细胞S-100强阳性,EnVision法 图5 Cyclin D1在RDD中呈特征性阳性,EnVision法:A.Cyclin D1在RDD病组织细胞胞核及胞质中~强阳性;B.Cyclin D1在RDD组织细胞胞核中等强度阳性;C.Cyclin D1在LCH表现为核强阳性;D.坏死性淋巴结炎增生组织细胞Cyclin D1阴性,仅散在血管内皮细胞阳性
2.3 免疫表型病变组织细胞单核/巨噬细胞相关抗原CD68阳性(图3),S-100呈强阳性(胞核+胞质)(图4),CD1a阴性;浆细胞CD38、CD138阳性;伸入的淋巴细胞CD3、CD5、CD20阳性;Ki-67增殖指数5%~30%,大部分为炎细胞阳性,病变组织细胞增殖指数均低于10%。RDD组织细胞Cyclin D1均阳性(Cyclin D1均中~强核阳性,个别病例组织细胞胞质弱阳性),对照组2例LCH表现为Cyclin D1核强阳性,2例组织坏死性淋巴结炎中增生组织细胞呈阴性,仅散在血管内皮细胞阳性(图5)。
2.4 分子检测对3例RDD的6个样本进行了基因检测。使用的参考基因组版本为hg19,报告中的变异命名遵从HGVS指南中的相关规定进行命名。共发现7种基因突变,包括MAP2K1、NTRK1、KRAS、FGFR3、TSC1、TSC2、NF2(表2)。

表2 3例RDD 6个样本分子检测结果
3 讨论
RDD是一种罕见的组织细胞性疾病,1969年Rosai和Dorfman对4例RDD进行了详细描述,并将其命名为“窦组织细胞增多症伴巨大淋巴结病”[1]。根据RDD病变累及范围可分为结内型、结外型及混合型[2]。
RDD主要影响儿童和年轻人,平均发病年龄20.6岁,男性略多于女性[3]。大多数患者以局部淋巴结无痛性肿大为主要或唯一表现而就诊,可伴发热、体重下降和盗汗等症状,病程长,病情进展缓慢,患者一般情况好[4]。40%患者可出现结外组织或器官受累[5]。本组12例男女比为2∶1,男性明显多于女性;患者平均年龄38.9岁,大于文献报道。本组66.7%出现结外受累(8/12),50%表现为单一结外受累(6/12),50%以发现肿块为主要症状就诊,50%以相应结外器官受累症状就诊。纵隔是RDD较为罕见的发病部位,既往仅见个例报道[6-8];本组1例上纵隔病例为39岁男性,伴有颈部淋巴结累及,该患者伴有反复发热症状,4年内住院治疗12次,最后一次入院才明确诊断,治疗效果差,明确诊断后半年去世。4例累及中枢神经系统的病例,1例合并有颅底骨质破坏伴鼻咽部累及,活检明确诊断后治疗缓解,随诊87个月无症状,1例患者切除后无复发,1例患者术后7个月后复发再次手术,术后1年无异常,1例会诊病例失访。文献报道皮肤型RDD更常见于老年妇女,平均发病年龄43.5岁[9];本组3例皮肤型RDD中2例为男性,平均年龄54.3岁。
RDD主要的病理形态学特征是低倍镜下“亮区”与“暗区”交替分布,高倍镜下病变组织细胞内可见淋巴细胞、浆细胞及红细胞“伸入现象”。在免疫表型上,病变组织细胞除表达单核/巨噬细胞相关抗原(CD68、CD163)外,胞核及胞质S-100阳性是RDD的重要诊断线索,淋巴结型RDD皮质内浆细胞可被浆细胞标志物(CD38、CD138和MUM1)染色,淋巴细胞表达CD3、CD5及CD20。
在鉴别诊断上,RDD一方面需与富于浆细胞的疾病鉴别。(1)与IgG4相关性疾病鉴别:后者纤维化更明显,且可出现闭塞性血管炎。(2)与浆细胞瘤鉴别:后者浆细胞呈膨胀性肿瘤性增生,浆细胞群体中淋巴细胞稀少,且不存在成片增生的组织细胞。(3)与浆细胞型Castleman病鉴别:后者淋巴滤泡反应性增生,滤泡周出现浆细胞,无组织细胞聚集成片的现象。另一方面需与富于组织细胞的疾病鉴别。(1)与LCH鉴别:LCH病变主要位于淋巴结的副皮质区,肿瘤细胞中等大小,细胞核可呈咖啡豆样外观,可见核沟,胞质较RDD少,且无炎细胞“伸入现象”,CD1a、CD207(Langerin)和S-100阳性提示克隆性增殖[10],无浆细胞显著增多现象。(2)与早期的组织细胞坏死性淋巴结炎鉴别:后者常坏死不明显,增生的组织细胞中可见少量吞噬现象,但组织细胞胞质不如RDD多,吞噬的为核碎裂成分,不是完整的淋巴细胞、浆细胞或红细胞,伴随的T淋巴细胞多,而浆细胞极少,但存在浆样树突细胞,CD123阳性,而CD38、CD138阴性等。(3)与Erdheim-Chester病鉴别:后者为黄瘤样组织细胞,胞质中常见脂质,并可见杜顿氏多核巨细胞,合并梭形细胞,且梭形细胞表达组织细胞抗原,可见淋巴细胞浸润,浆细胞极少见,且约60%病变出现BRAF V600E基因突变。(4)与黄色纤维组织细胞瘤鉴别:后者无成片增生的浆细胞,组织细胞胞质中常见脂质空泡。(5)与黏液变的神经鞘瘤鉴别:后者无浆细胞聚集现象,黏液变区域与RDD的组织细胞区域在高倍镜下明显不一样,淋巴细胞浸润极少。(6)与肉芽肿性疾病鉴别:后者组织细胞聚集呈团,黏附性强,细胞胞质相对RDD细胞胞质少,且无“伸入现象”。除梅毒等特殊肉芽肿外,浆细胞聚集现象非常少见。
RAS-RAF-MEK-ERK细胞信号通路在实体肿瘤和血液系统恶性肿瘤中发挥重要作用。该通路能影响几种不同的细胞功能,包括增殖、凋亡、血管生成、迁移和存活[11]。既往在对LCH发病机制的分子研究中发现了与MAPK/ERK途径相关的互斥复发性体细胞突变,包括BRAF V600E和MAP2K1[12],在Erdheim-Chester病中也有报道[13]。RDD的病因尚不明确,长期以来被认为是非克隆疾病,但近年来部分支持RDD克隆性的证据已被发现。Garces等[14]的研究显示:高达33%的RDD病例显示KRAS或MAP2K1突变,这表明部分病例可能是克隆性的。Durham等[15]对17例RDD进行靶向DNA/RNA测序和全外显子测序并发现了KRAS、MAP2K1、NRAS及CSF1R突变。本文对3例RDD进行NGS检测,发现了MAP2K1、NTRK1、KRAS、FGFR3、TSC1、TSC2、NF2突变。例1为56岁男性,皮肤RDD,MAP2K1基因3号外显子错义突变,c.371C>A(p.P124Q)以及NTRK1基因9号外显子突变,c.1024G>T(p.A342S)。MAP2K1基因的c.371C>A(p.P124Q)导致基因编码蛋白第124位氨基酸由脯氨酸转变为谷氨酰胺,在一项临床研究中[16],1例携带MAP2K1 P124Q突变的Erdheim-Chester病患者使用cobimetinib治疗后疗效为完全缓解。NTRK1基因的c.1024G>T p.A342S导致基因编码蛋白第342位氨基酸由丙氨酸转变为丝氨酸,其与肿瘤发生发展的相关性暂不明确。例2为19岁男性,中枢神经系统RDD,KRAS基因4号外显子错义突变,c.351A>C(p.K117N)。KRAS基因是人体内最为常见的原癌基因,其负责编码的RAS蛋白在细胞内的信号通路中起着信号转导作用,KRAS突变在多种肿瘤中均有发生,在肺癌中占15%~25%[17],在结直肠癌中高达40%[18],在胰腺癌中甚至高达70%[19]。KRAS p.K117N属于明确的致病性突变,该突变的发生可导致KRAS蛋白及其下游MAPK信号通路持续激活,从而促进肿瘤的发生与发展。例3为66岁女性,皮肤及腋下淋巴结RDD,发现多个基因突变:FGFR3基因13号外显子错义突变,c.1949A>C(p.K650T);TSC1基因15号外显子错义突变,c.1460C>G(p.S487C);TSC2基因34号外显子错义突变,c.4270G>C(p.D1424H);NF2基因13号外显子错义突变,c.1439C>T(p.T480M)。FGFR3隶属FGFR酪氨酸激酶家族,参与细胞的增殖、生长、迁移与分化。FGFR3突变在多种肿瘤中均有报道,包括乳腺癌、结直肠癌、肺癌、胰腺癌、肾细胞癌、膀胱癌等。多篇文献报道显示[20-22],胚系FGFR3 K650T突变与家族性软骨发育低下和黑棘皮症相关,OncoKB数据库记录该突变为疑似致病性突变。本例属于首次在RDD中发现FGFR3 K650T突变。TSC1、TSC2、NF2突变在既往RDD中未见报道,它们对临床用药意义尚不明确。
Cyclin D1是MAPK通路的下游标记之一。92.3%~100%的LCH病例发现了Cyclin D1阳性,与MAPK普遍激活保持一致[23-24]。多篇文献报道[25-27]Cyclin D1在几乎所有RDD中均阳性,且显著高于反应性组织细胞增生和IgG4相关性疾病。本组9例RDD行Cyclin D1免疫组化染色,8例出现病变组织细胞核中~强阳性,部分病例组织细胞胞质亦阳性。Cyclin D1在RDD中特征性阳性,加强了RDD是肿瘤性病变的证据,不仅提示MAPK通路激活可能参与RDD的发病机制,也能作为诊断RDD的一个重要辅助标记。
RDD通常具有自限性且预后良好,但局部切除后容易复发[28]。本组有随诊资料的9例患者中4例复发。文献报道部分患者可能死于并发症或感染[3],特殊部位反复复发也容易致命。对于没有并发症和无症状的皮肤RDD可观察随访,手术治疗是单灶结外RDD和有症状RDD的首选治疗方式,手术不能完全切除的多灶RDD需辅以系统治疗[4-5]。RDD克隆性证据的发现为使用靶向药物治疗提供了理论依据,目前共识建议伴MAPK通路相关基因突变的严重或难治性RDD可考虑行靶向治疗[4]。
总之,RDD是一种有具有独特病理形态学特征,通常具有自限性且预后良好的组织细胞疾病。组织细胞标记(CD68、CD163)与S-100在组织细胞中联合表达,Cyclin D1阳性也可作为RDD辅助诊断的有用标记,伴发的浆细胞增多虽然目前意义不明,但具有重要的鉴别诊断意义。目前RDD尚未被归类为肿瘤性疾病,但一些支持克隆性证据改变了我们对这类疾病的理解。激素治疗往往有效,难治性病例和特殊部位病例也存在致命性可能。
