脑海绵状血管畸形KLF2、KLF4信号通路的研究进展
2022-10-22峰综述利审校
张 峰综述 刘 利审校
脑海绵状血管畸形(cerebral cavernous malformations,CCM)是由桑椹样扩大的不规则血管形成的,缺乏平滑肌、弹性组织和完整的基底膜,因此血管壁薄,易渗漏[1]。CCM的发病率约为0.5%,以中枢神经系统多见,导致头疼、中风、癫痫发作和局灶性神经功能缺失。CCM 发病形式包括散发性CCM(sporadic CCM,SCCM)和家族性CCM(familial CCM,FCCM),其中FCCM 与染色体7q 的CCM1(KRIT1)、染色体7p 的CCM2(OSM)和染色体3q 的CCM3(PDCD10)基因有关。研究发现,参与CCM 损伤的信号通路有:MEKK3-Krüppel样因子(Krüppellike factor,KLF)2/4、RhoA/ROCK、内皮-间充质转化(endothelial-to-mesenchymal transition,EndMT)、血小板反应蛋白1(thrombospondin 1,TSP1)丢失、促炎状态、氧化应激和自噬[2,3]。本文主要介绍KLF2、KLF4在CCM信号通路中所起的作用。
1 KLF2、KLF4结构和功能
KLF家族是真核生物体内一类结构高度保守的转录因子,其C-末端有3个连续高度保守的C2H2锌指结构域,通过结合靶基因启动子GC 富含序列,调控相应基因的表达;其N-末端属于转录调控结构域,高度变异,通过结合特异蛋白质,介导多种因子的转录。KLF4在人体内表达广泛,主要存在于细胞核内,其特征是:因作用靶基因的不同,具有转录活化或转录抑制的双重调节作用。KLF4 通过对其下游靶基因的调控,参与细胞的增殖、分化、凋亡以及血管生成和肿瘤的发生发展[4]。KLF2可受血流切应力的诱导,调节内皮细胞多种功能基因的表达,参与调节炎症、凝血、血管舒缩和血管生成等多种过程[5]。KLF2和KLF4之间有许多共同的靶点,但它们对共同调控的启动子在亲和力方面存在个体差异,因而表现出特异性。Zhou 等[6]发现,CCM 小鼠模型的早期形成与KLF2、KLF4的过度表达和Rho/ROCK活性的增加有关。当CCM 蛋白缺陷后,激活MEKK3-MEK5-ERK5-MEF2 信号轴,使转录因子KLF2、KLF4 表达增加[7],KLF2 可进一步激活下游RhoA/ROCK信号通路,而KLF4则激活其下游的EndMT信号通路[8],KLF2 与KLF4 二者也可共同作用于TSP1,使其表达减少,从而促进CCM的发展及其出血性演变[9]。目前认为,KLF2、KLF4 是CCM 早期发展和进展的关键,可以作为CCM发病机制的研究重点。
2 信号转导
2.1 KLF2/4 与MEKK3-MEK5-ERK5-MEF2 信 号 轴有丝分裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路表达于所有真核生物内,是将细胞上接收的刺激信号传递到相应蛋白所必需的信号通路。因此,MAPK通路在细胞的炎症反应、分化、生长和增殖等过程中起着非常重要的作用。MAPK 激酶激酶3(MAPK kinase kinase 3,MEKK3)是MAPK 信号通路中重要的节点,通过磷酸化激活细胞外信号调节激酶激酶5(Mitogen/extracellular signal-regulated kinase kinase 5,MEK5),进一步激活细胞外信号调节蛋白激酶5(extracellular signal-regulated protein kinase 5,ERK5)从而发挥生物学效应。Maddaluno 等[7]发现,人体内三种CCM蛋白相互作用形成的蛋白质复合物(CCM signaling complex,CSC)是MEKK3 级联反应的抑制因子。当CSC 蛋白缺陷后,会激活MEKK3,激活MEKK3-MEK5-ERK5信号途径。磷酸化ERK5可以通过调节肌细胞增强因子2A、2C的转录活性,进一步促进KLF2、KLF4的表达[8,9]。Cuttano等[10]发现,敲除KLF4基因表达可显著降低CCM1小鼠病死率,并适度改善血管病变。
2.2 TLR4、CDC42 与MEKK3-KLF2/4 Toll 样受体4(toll like receptors 4,TLR4)可识别病原微生物细胞壁上的保守成分脂多糖(lipopolysaccharide,LPS)。LPS作为Ⅰ型急性期反应蛋白可诱导机体产生炎症免疫反应。TLR4也是一种保守的Ⅰ型跨膜蛋白,可以将细胞外刺激传递到细胞内,进而通过信号转导参与多种疾病发生发展过程。Tang 等[11]在CCM1 小鼠模型中发现,诱导革兰氏阴性细菌脓肿,会显著增加CCM病变表型的严重程度,提示TLR4可能是MEKK3 的上游靶点。当革兰氏阴性细菌产生LPS 后,可被TLR4 蛋白识别,进一步通过TLR4-MEKK3-KLF2/4信号途径来加速CCM的形成。
细胞分裂周期蛋白42(cell division cycle 42,CDC42)是Ras 超家族GTP 酶中的核心成员,位于细胞质,包含191个氨基酸,主要参与调解细胞骨架中肌动蛋白重排与调控蛋白激酶,并通过在与GTP 结合/活化状态和与GDP 结合/失活状态之间循环切换,从而在复杂的细胞信号转导网络中起到开关作用。当CDC42 功能丧失后,会损害脑内皮细胞的发芽、分支形态发生、轴向极性以及其在脑组织中的正常分布。Castro等[12]发现,CDC42也参与CSC之间的相互作用并形成复合物,当脑内皮细胞CDC42 缺失后,也通过激活MEKK3-MEK5-ERK5 信号通路,进而导致KLF2、KLF4的过表达,诱导CCM的形成。
2.3 KLF2 与RhoA/ROCK 信 号 通 路RhoA 是 一 种 小的GTP 结合蛋白,具有高度保守的GDP/GTP 结合区、GTP酶活性区、靶区和膜定位结构。作为一种分子开关,RhoA 的主要功能是调节细胞骨架重组、细胞生长和基因表达,在细胞信号转导通路中起着重要的作用。RhoA 的功能主要是通过ROCK实现的,ROCK有ROCK1和ROCK2两种异构体,在激酶结构域的水平上有65%的总体同源性和92%的相似性。Richardson等[13]发现,当体内出现任何一种CCM蛋白缺陷,都会激活MEKK3-KLF2-RhoA-ROCK1/2-pMLC 信号通路,最终导致肌动蛋白应激纤维的增加、细胞黏附性的降低以及血管通透性增加;当抑制KLF2 表达时,CCM1 小鼠模型中病变表现出99%的恢复,且组织学上只有少量的小静脉扩张。
2.4 KLF4 与EndMT EndMT 被认为是上皮间质转分化(epithelial-to-mesenchymal transition,EMT)的一种亚型,是指内皮细胞失去其自身特性而获得间质细胞和干细胞样特性的表型转变过程。EndMT的特点是细胞连接结构的改变、细胞极性丧失,细胞增殖或迁移增加,最终导致血管生成增加,并可在炎性微环境下形成桑椹状[14]。在正常人体内,CSC可通过抑制小GTP酶RAS相关蛋白1的效应器来控制内皮细胞屏障功能。在CCM蛋白缺陷的情况下,脑内皮细胞间粘附连接减少,可诱发内皮细胞经历EndMT 形成CCM。EndMT 主要是由内源性骨形态发生蛋白(bone morphogenetic protein,BMP)6 上调介导的,可激活EndMT 发生有关的BMP 和转化生长因子-β(transforming growth factor-β,TGF-β)信号通路[7]。Zhang 等[15]发现,KLF4 是CCM 蛋白缺陷后产生End-MT 的关键。BMP6 作为KLF4 的靶基因,KLF4 可以通过诱导BMP6 启动子的活性,从而诱发EndMT 的形成。研究报道,KLF4除诱导BMP6外,还可以直接通过与某些EndMT 标志物启动子的结合来诱导EndMT 的产生,如干细胞抗原1(stem cell antigen 1,SCA1)和成纤维细胞特异性蛋白(fibroblast-specific protein 1,FSP1)。同时,在CCM1 小鼠模型中,KLF4基因敲除既会显著降低EndMT典型标志物(DNA结合抑制因子1、FPS1、SCA1)中mRNA与蛋白水平,又会导致与EndMT发生有关的BMP6被强烈抑制。
2.5 KLF2/4 与TSP1 TSP1 是一种细胞基质糖蛋白,可通过调节细胞增殖、黏附和凋亡,来维持细胞平衡;同时,TSP1 也是最有效和最具特征性的内源性血管生成抑制剂之一,在血管生成过程中表达上调,通过抑制体内基质金属蛋白酶的表达来抑制血管生成,从而限制血管密度。Lopez-Ramirez 等[8]发现,CCM 蛋白缺失后会引起KLF2 和KLF4 的过表达,从而导致其下游TSP1 的表达被抑制。TSP1 可以促进血管内皮细胞生长因子受体2(vascular endothelial growth factor receptor 2,VEGFR2)降解,从而抑制VEGFR2 信号通路。当TSP1 表达减少时,会增强VEGFR2 信号通路,从而导致CCM 的发生。VEGFR2 也与内皮细胞有丝分裂和血管生成密切相关。当VEGFR2活化后,可增加参与内皮细胞渗漏、应力纤维形成和血管生成密切相关的血管内皮钙粘蛋白和Beta连环蛋白磷酸化和迁移,从而诱发CCM病变形成[16]。Lopez-Ramirez 等[8]在用KLF2、KLF4来抑制TSP1表达的实验中发现,当诱导人内皮细胞异位表达KLF2 和KLF4 后,TSP1 表达水平与CCM1 沉默后的水平相似;并发现KLF2 和KLF4 的过表达也可以导致TSP1 mRNA表达水平下降。
综上所述,KLF2、KLF4 被MEKK3-KLF2/4 信号通路激活后,在EndMT、RhoA/ROCK 信号通路以及TSP1 表达中起到重要的桥梁作用(图1),是CCM 发生发展的关键。在CCM小鼠模型中,KLF2、KLF4基因失活可大大减少CCM 病变的形成,因此,KLF2、KLF4 可作为未来CCM 发病机制信号通路的研究重点。KLF2、KLF4作为新的药物治疗靶点,也可为不可触及的病变提供新的治疗方案,如脑干CCM、多发性CCM或位于功能区的病变。
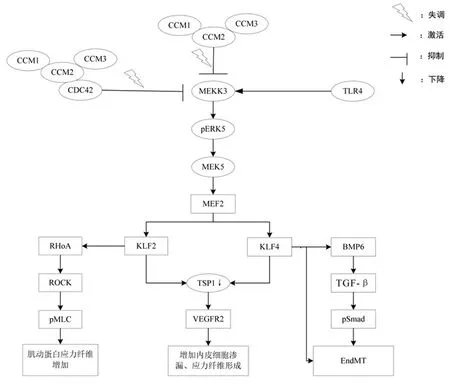
图1 CCM 发生发展过程中与KLF2/4 有关信号通路图
