猪伪狂犬病病毒流行毒株JZ-45的分离鉴定与变异分析
2022-10-21苗信永孔令昊赵世杰崔志莹徐朋丽张宜娜李新生夏平安
苗信永,孔令昊,陈 雨,赵世杰,崔志莹,李 闻,徐朋丽,陈 静,张宜娜,李新生,夏平安*,吴 斌
(1.河南农业大学 动物医学院,河南 郑州 450002;2.华中农业大学 动物医学院,湖北 武汉 430070)
伪狂犬病(pseudorabies,PR)是由伪狂犬病病毒(pseudorabies virus,PRV)引起的一种高度接触性传染病,该病没有特异性宿主,可感染猪、熊、牛、羊等多种哺乳动物[1],感染后主要引起动物发热、奇痒(猪除外)以及脑脊髓炎等临床症状[2]。自1902年在匈牙利首次发现PRV以来,该病毒一直被认为是最重要的病原体之一[3]。1947年,李树清等[4]首次发现猫PRV,此后该病在我国不同物种间的传播被广泛记录。猪作为该病毒的唯一自然宿主,是PRV最主要的传播媒介[5],并且每个阶段的猪均可感染PRV,感染后的猪普遍表现为体温升高,但仔猪主要以神经症状为主,病死率高达100%;育肥猪感染PRV后没有明显的症状,基本为隐性感染,但可长期携带病毒,成为猪场最主要的传染源;妊娠母猪感染PRV后会引起流产、死胎以及呼吸系统疾病;公猪感染后会引起繁殖障碍[6]。20世纪60-90年代,PRV在我国不少地区一直存在,并有流行的趋势,严重阻碍了我国养猪业的发展。近年来,随着我国养猪业规模的不断发展以及生猪饲养市场之间的频繁交流,PR在我国范围内开始广泛流行,给我国养猪业造成了巨额损失[7]。虽然我国通过广泛应用从匈牙利引进的商业疫苗Bartha-K61(一种基于PRV Bartha-K61株的减毒活疫苗),使得PR在我国得到了很大程度地控制[8]。但2011年以后,在我国使用过商品化PRV疫苗的规模化养猪场再次出现了与PR极为相似的疫病,导致妊娠母猪产死胎、流产,仔猪患有神经症状导致其死亡[9]。后经我国学者证实,该病是由PRV的变异株暴发引起的[10]。本试验利用PCR检测方法对2018—2020年河南省部分地区的疑似PRV病料进行检测,所获得的阳性样品进行病毒分离鉴定,获得1株PRV毒株JZ-45,进行核苷酸序列对比分析等研究,从遗传变异方面解释现阶段流行毒株与2011年变异毒株的亲缘关系,以期为中国PRV的流行病学以及新型PRV疫苗研究提供参考资料。
1 材料与方法
1.1 样品来源2020年间河南省开封、周口、信阳、焦作、新乡、济源、登封等地区规模化养猪场送检的临床样品。
1.2 试验动物及细胞健康新西兰白兔2只,购自郑州幸福养殖场;4~6周龄SPF级昆明雌鼠30只,购自郑州大学实验动物中心;猪睾丸细胞(ST),由河南农业大学动物医学院分子免疫学实验室保存。
1.3 主要试剂RPMI-1640培养基购自上海克隆生物化学有限公司;胎牛血清购自杭州四季青生物工程有限公司;高保真DNA聚合酶购自南京诺唯赞生物科技有限公司;病毒DNA提取试剂盒购自生工生物工程(上海)股份有限公司。
1.4 引物设计参照GenBank收录的PRV全基因序列设计PRV gB、gE检测引物;gE、gC全基因引物,用以临床病料的检测、病毒核酸鉴定以及病毒变异性分析(表1)。引物均由生工生物工程(上海)股份有限公司合成,具体信息见表1。PCR扩增反应程序为94℃ 5 min;98℃ 30 s,58℃ 30 s,68℃ 2 min,35个循环;72℃ 延伸10 min。
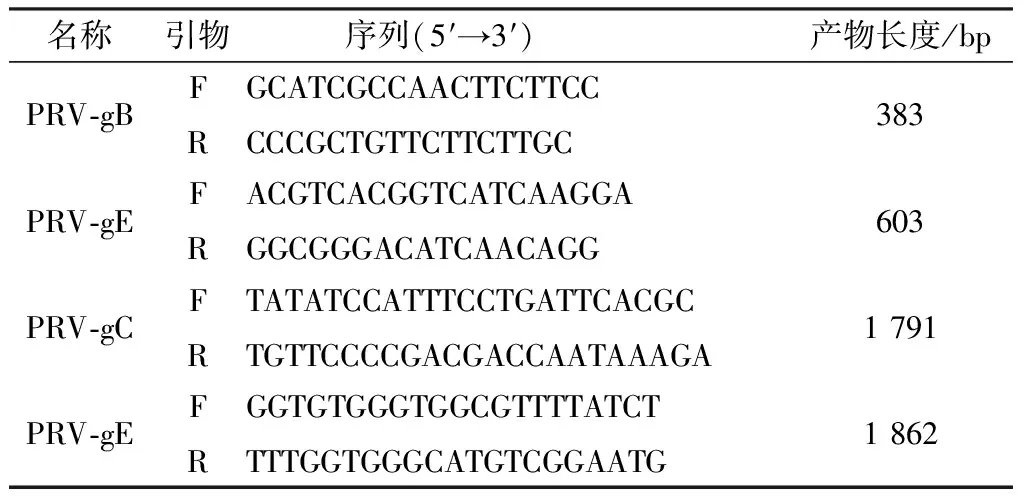
表1 引物序列及扩增长度
1.5 病毒分离培养取适量阳性组织样品,剪碎、研磨、匀浆、反复冻融3次后取上清,用0.22 μm无菌滤器过滤。按1 mL/25 cm2接种于汇合度至80%左右的ST细胞,盲传2~3代后,ST细胞出现稳定且典型CPE,待80%的细胞出现病变时,收集备用。
1.6 空斑纯化病毒液进行10倍倍比稀释(10-1~10-5)后,接种于6孔板内汇合度至80%左右的ST细胞,500 μL/孔,于37℃、5% CO2细胞培养箱中作用1 h后,加入含2%血清和1%低熔点琼脂糖的细胞培养基,室温冷却至凝固后,倒置放入细胞培养箱待出现CPE现象后,使用枪头挑取空斑置于培养液中,反复冻融3次后,用RPMI-1640稀释并继续接种ST细胞和继续进行下轮的空斑纯化。经过6轮空斑纯化,进行扩大培养。
1.7 间接免疫荧光试验(IFA)将病毒液接种于6孔板中单层ST细胞中,同时设空白对照,12 h左右未出现明显CPE时,弃上清,加入1.5 mL的细胞组织固定液,固定30 min弃掉,加入1.5 mL 5%的BSA,封闭2 h弃去,加猪抗PRV多抗,37℃孵育1 h弃掉,加入FITC标记的兔抗猪IgG,37℃孵育1 h,PBS洗涤,于倒置荧光显微镜下观察。
1.8 病毒TCID50测定将病毒液10倍倍比稀释(10-1~10-10)后,接种于96孔板内汇合度至80%左右的ST细胞,于37℃、5% CO2细胞培养箱中作用1 h后,加入细胞维持液继续培养96 h,按照Reed-Muench法计算病毒滴度TCID50。
1.9 病毒LD50的测定及动物试验将病毒液10倍倍比稀释(10-1~10-5)后,于腹股沟皮下注射小鼠,100 μL/只,每组5只,对照组注射等量RPMI-1640。逐日观察小鼠状态及记录小鼠的死亡数量,对死亡小鼠进行剖检制作组织病理切片,观察心脏、肝脏、脾脏、肺脏、肾脏、脑等组织的病理变化。根据Reed-Muench法计算PRV JZ-45分离毒株的LD50。同时将100×LD50病毒液接种家兔,500 μL/只,每天观察家兔的健康状态,记录出现的症状及病死时间。
2 结果
2.1 PRV核酸鉴定将送检的临床样品研磨,离心抽取上清,用DNA提取试剂盒提取病毒基因组DNA,用gB、gE基因双重PCR鉴定。凝胶电泳在383,603 bp左右出现特异性条带,样品扩增结果见图1。
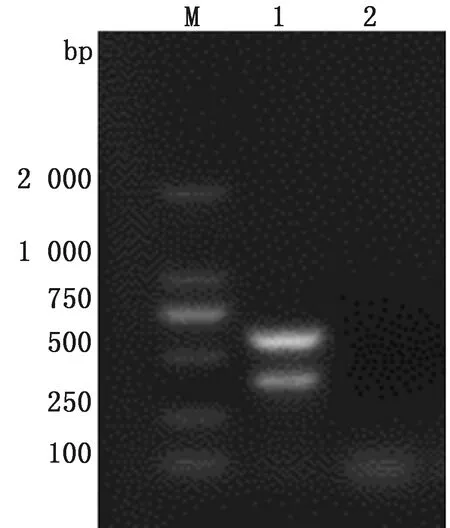
M.DL2000 DNA Marker;1.送检样品;2.阴性对照
2.2 病毒分离培养将上述检测为阳性的PRV病毒液,接种ST细胞,10~12 h后,细胞折光性增强出现亮点,且细胞变大变圆、细胞脱落、拉网、破碎(图2)。
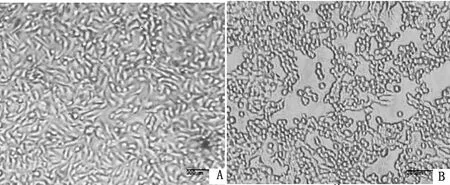
A.正常的ST细胞;B.感染PRV病变的ST细胞
2.3 病毒空斑纯化将病毒液接种细胞36~48 h后,在倒置的6孔板中可看到明显的空斑(图3),待空斑长至一定大小后,从中挑取单个空斑,经过连续纯化6轮,获得纯化的病毒株,命名为JZ-45。
A.PRV接种ST细胞空斑形态;B.阴性对照
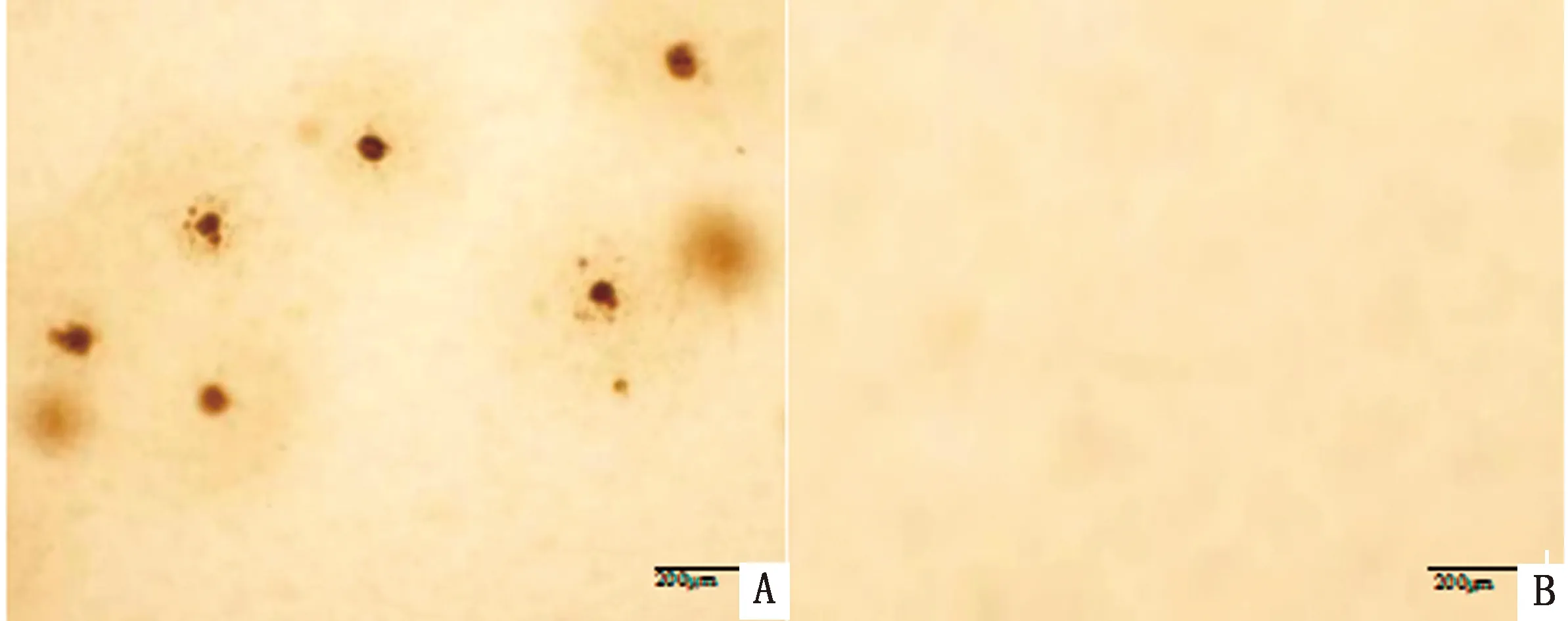
2.4 间接免疫荧光试验分离的JZ-45毒株接种ST细胞,将猪抗PRV阳性抗体做100倍稀释,后用FITC标记的兔抗猪抗体与一抗结合,进行免疫荧光鉴定。结果显示,在荧光显微镜下可以观察到细胞出现荧光标记的细胞(图4),进一步证明该分离株为PRV。
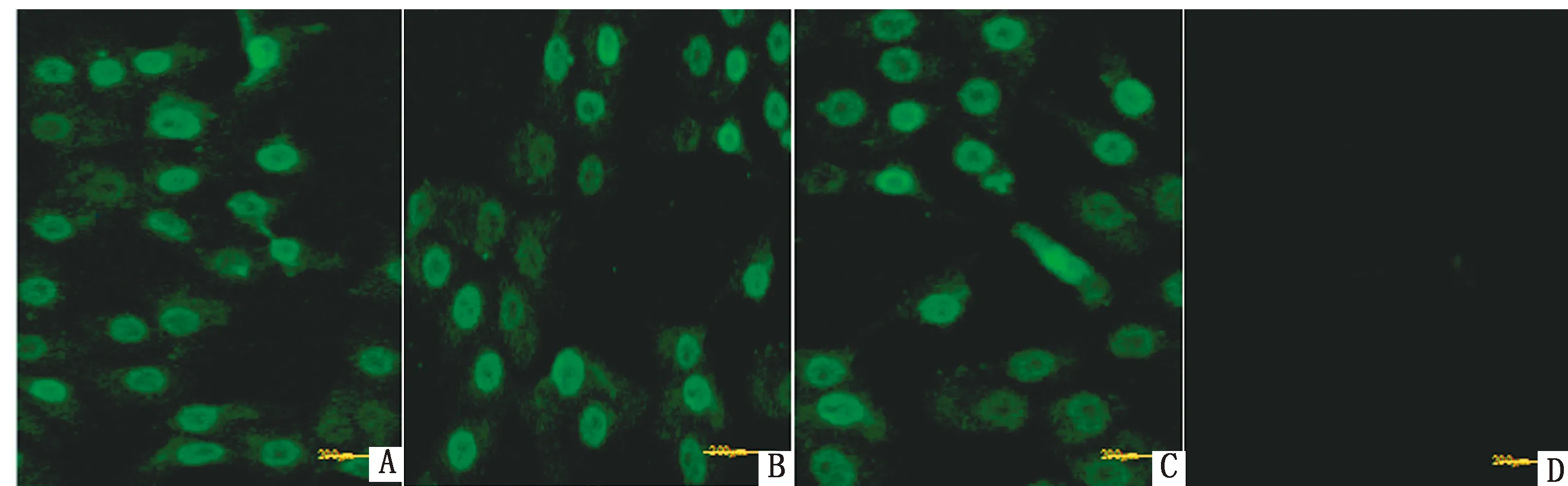
A,B,C.PRV接种ST细胞;D.空白对照
2.5 病毒TCID50测定PRV JZ-45毒株接种ST细胞72 h后,观察每孔CPE,按Reed-Muench方法计算其TCID50为10-6.58/0.1 mL。
2.6 病毒LD50测定及组织病理学观察PRV JZ-45毒株接种小鼠3 d后出现PRV典型症状:奇痒、流涎,啃咬接种部位,致使皮肤出血,记录小鼠病死数量,持续观察14 d,根据Reed-Muench法测得病毒LD50为10-2.681/0.1 mL;对测LD50过程中死亡小鼠剖检制作组织病理切片,观察其组织病理变化结果(图5)。
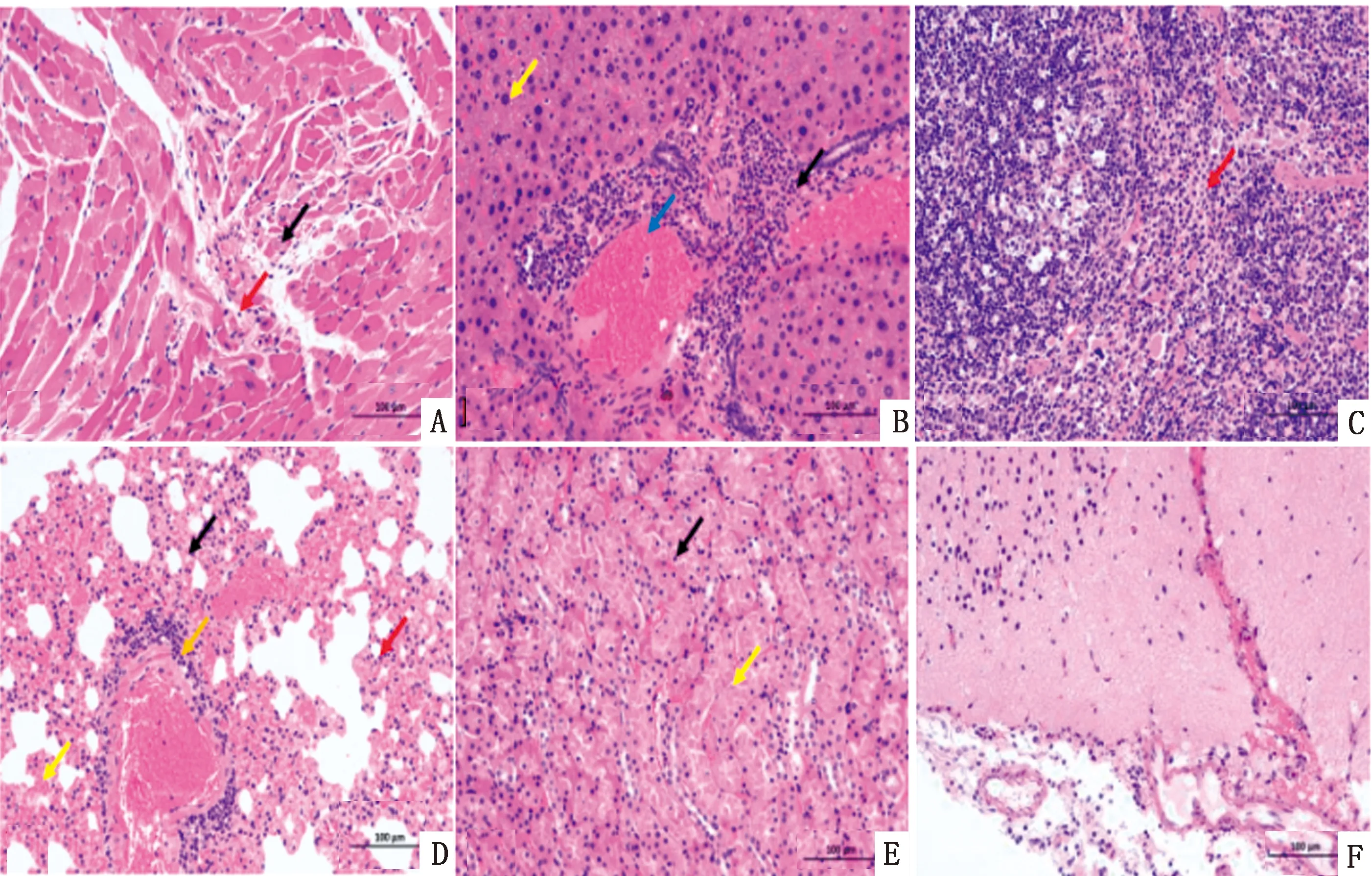
A.心肌纤维溶解(黑色箭头),点状炎性细胞浸润(红色箭头);B.肝脏有炎性细胞灶性浸润(黑色箭头),胞质内有空泡(黄色箭头);C.红髓中有中性粒细胞浸润(红色箭头);D.肺组织肺泡壁增厚(黑色箭头),炎性细胞浸润(红色箭头),肺泡壁毛细血淤血(黄色箭头),少量炎性细胞浸润(橙色箭头);E.肾脏组织肾小管上皮细胞坏死脱落(黑色箭头),肾小管管腔内可见脱落的坏死细胞碎片(黄色箭头);F.脑部有炎性细胞点状浸润
2.7 动物试验将该病毒液接种家兔后,1 d内无明显临床症状,2 d时,体温升高,且食欲不振、3 d 时出现四肢痉挛、身体蜷缩,且对注射部位抓挠啃咬,注射部位脱毛、皮肤撕裂出血,并死亡。
2.8 PRV gC、gE基因扩增将分离所得毒株用DNA提取试剂盒提取病毒基因组DNA,用PRV gC、gE全基因特异性引物扩增。产物经凝胶电泳在1 791,1 862 bp左右出现特异性条带,样品扩增结果见图6。
2.9 病毒分离株变异性分析基于PRV分离株gC、gE基因推导氨基酸序列,利用MEGA 7.0软件绘制其遗传进化树。结果如图7,8所示,PRV gC、gE基因遗传进化树将参照毒株分为2个基因型,即Group 1和Group 2型:Group 1型均为国内毒株,且Group 1型可进一步分为2个基因亚型,分别是以HN1201为代表的变异毒株组成的基因亚型Group1.1和以Ea为代表的国内经典毒株组成的基因亚型Group 1.2。Group 2型均为美洲和欧洲毒株。由图6,7可知分离得到的PRV毒株JZ-45 gC、gE基因与国内经典毒株(LA、Ea、Fa、NIA3、LXB6株等)均位于不同的进化分支,而与国内变异毒株亲缘关系更近。经核酸序列分析gC基因与国内变异毒株GDWH、DL14、HN1201等同源性为99.9%~100.0%,与国内经典毒株Ea、Fa等同源性为99.2%~99.7%,与国外经典毒株NIA3、Kaplan等同源性为96.0%~96.2%,氨基酸序列对比表明,与国内变异毒株相比,氨基酸没有发生变异或缺失,与国内经典毒株Ea毒株相比,有4处变异:T34N、E99K、G194E和S233R,与国外经典毒株NIA3相比,在63位氨基酸位点处连续插入7个氨基酸(AAASTPA),并且gC全基因序列中存在30处氨基酸替换。经核酸序列分析gE基因与国内变异毒株CH、HeN1、HN1201等同源性为99.7%~99.8%,与国内经典毒株Ea、SC等同源性为96.7%~99.4%,与国外经典毒株NIA3、Kaplan等同源性为95.3%~97.8%,氨基酸序列对比表明,与国内变异毒株相比,氨基酸没有发生变异或缺失,与国内经典毒株Ea相比,有7处变异:G54D、Y286F、A308P、P403A、V448T、G510S和S518P以及2处氨基酸插入第447位(A)和492位(DG),与国外经典毒株NIA3相比,有27处氨基酸发生替换,在492位氨基酸位点处连续插入2个氨基酸(DG)。
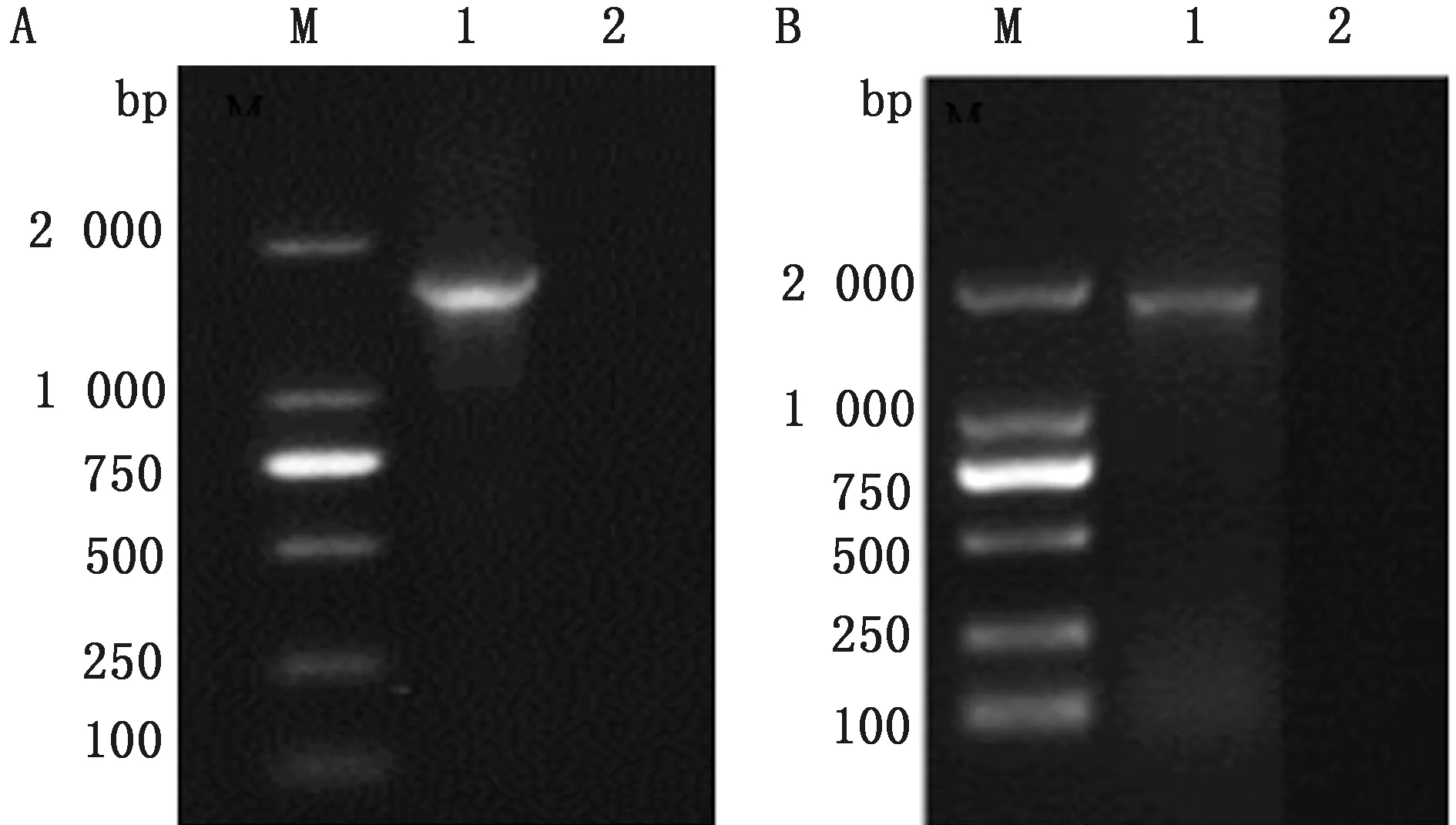
M.DL2000 DNA Marker;1.PRV JZ-45;2.阴性对照
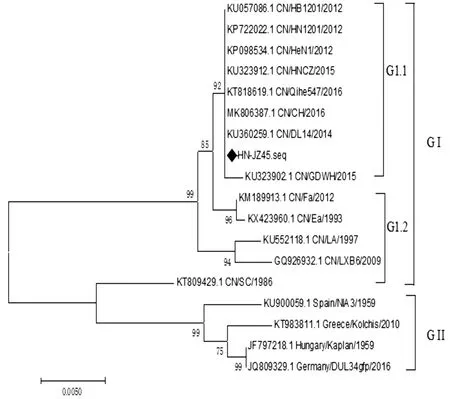
图7 PRV JZ-45株gC基因遗传进化树
3 讨论
PR是一种在世界范围内广泛流行,且多种动物均可感染的高度接触性传染病[11]。自我国首次报道PR以来,PRV便成为了危害我国养猪业最重要的病原之一[12]。2011年末,PRV变异毒株的出现,使得市场上传统PR疫苗的保护力度大大下降,规模化养猪场再次出现了PR流行趋势[13-14]。尤其在2017—2018年,PRV病原检测阳性率在河南省规模化养猪场有所上升[15]。本试验通过ST细胞培养、空斑纯化、动物接毒、间接免疫荧光等分离鉴定方法,从PCR检测为阳性的组织样品中分离获得1株可在ST细胞上增殖,且具有较强毒力的变异毒株PRV JZ-45。
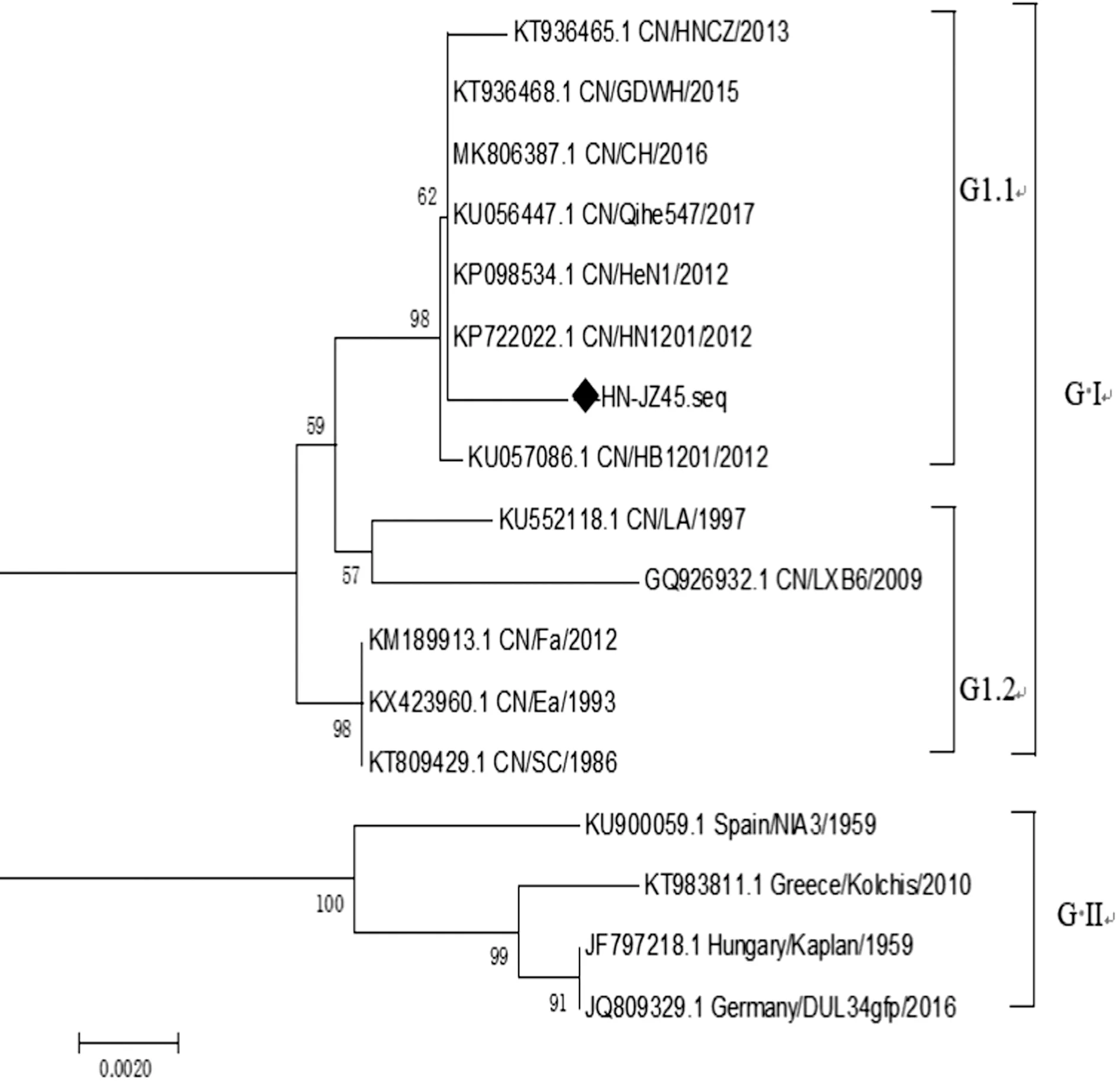
图8 PRV JZ-45株gE基因遗传进化树
PRV的基因组为线状双股DNA,全长约150 kb,相对分子质量约9.2×104kDa,编码70~100种蛋白,而影响病毒毒力强弱的主要是毒力基因编码的毒力蛋白,包括TK、gC、gE等[16-17],gC蛋白属于Ⅰ型膜蛋白,全长1 500 bp,共编码了479个氨基酸。该蛋白与病毒吸附和诱导机体产生中和抗体方面起到至关重要的作用[18]。gC蛋白的缺失会影响病毒的感染力,因此gC蛋白也是我国学者研究猪PRV疫苗的重要蛋白。本研究测序的PRV JZ-45毒株经核酸序列分析gC基因与国内变异毒株GDWH、DL14、HN1201等同源性为99.9%~100.0%,氨基酸序列没有发生变异,与国内经典毒株Ea毒株相比同源性为99.2%~99.7%,氨基酸序列有4处变异:T34N、E99K、G194E和S233R,与国外经典毒株NIA3、Kaplan等同源性为96.0%~96.2%,且在63位氨基酸位点处连续插入7个氨基酸(AAASTPA),表明近年来的PRV变异毒株正在向着远离经典毒株的方向发生变异,这也可能进一步解释了2011年末已免疫过Bartha-K61减毒苗的规模化猪场再次出现PRV感染流行的状况。gE基因是PRV主要毒力基因之一,全长1 737 bp,共编码了577个氨基酸。其编码的蛋白属于典型的囊膜蛋白,该蛋白可介导病毒在细胞与细胞之间的扩散以及病毒粒子的释放[19]。但gE蛋白的缺失不影响PRV病毒的复制,却可以显著影响PRV的毒力[20],因此,gE蛋白成为了人们研究减毒苗的关键蛋白之一。本研究测序的PRV JZ-45毒株经核酸序列分析gE基因与国内变异毒株CH、HeN1、HN1201等同源性为99.7%~99.8%,氨基酸没有发生变异或缺失,与国内经典毒株Ea、SC等同源性为96.7%%~99.4%,与国外经典毒株NIA3、Kaplan等同源性为95.3%~97.8%,与国内经典毒株Ea相比,有7处变异:G54D、Y286F、A308P、P403A、V448I、G510S和S518P以及2处氨基酸插入第447位(A)和492位(DG),有报道证明第48位和492~496位天冬氨酸(D)插入是鉴别PRV毒株是否为变异毒株的重要特征[21-22]。同时也进一步证实了分离得到的PRV JZ-45毒株为变异毒株。
据报道,引起PRV毒株变异的主要原因可能是某些规模化养猪场注射不同基因缺失减毒苗导致的不同疫苗毒株之间发生重组,使其毒力返强[23],现如今,变异毒株的流行,仍然对我国养猪业造成了巨大的经济损失,而PRV JZ-45变异毒株的分离,为下一步变异毒株的研究和相关疫苗的制备奠定基础。
