染色质转座酶可及性测序及其在木本植物中的应用前景
2022-10-19王子玥刘光欣席梦利
王子玥,甄 艳,刘光欣,席梦利
(南京林业大学,南方现代林业协同创新中心,林木遗传与生物技术省部共建教育部重点实验室,江苏 南京 210037)
真核生物的染色体由DNA分子绕组蛋白八聚体1.75圈形成的核小体结构进一步折叠形成[1]。通常情况下组蛋白八聚体上缠绕的DNA(core DNA)不能与转录因子(transcription factor, TF)结合,而位于核小体之间的DNA(linker DNA)因未与核心组蛋白缠绕而处于相对裸露状态[2-3],这些裸露区域有利于TF结合,该区域被称为可及性染色质区域(accessibility chromatin regions, ACRs)或开放染色质(open chromatin)。真核生物可及性染色质区域占总基因组 DNA 序列的 2%~3%,且该区域超过 90%与转录因子的结合相关。染色质这种允许其他调控因子结合的特性称为染色质可及性(chromatin accessibility)[4]。植物作为固着生物,可以通过形态、生理和生化过程的调控等多种方式来适应环境的变化[5],其中转录水平的调控是最主要的调控方式。只有当转录因子与相应的顺式调控元件(cis-regulatory elements, CREs)相互作用才能有效调控转录,CREs一般包括启动子、增强子、抑制子等[6]。因此,研究染色质可及性区域对转录调控元件的鉴定、转录因子结合位点的识别及基因表达调控的解析等方面都具有重要意义。
染色质可及性区域因其裸露特性,对DNase Ⅰ和Tn5转座酶都表现出高度敏感性。研究者最初采用Southern 杂交技术鉴定DNase Ⅰ的高敏感位点(DNase Ⅰ hypersensitive sites, DHSs),但该技术费时费力,且只适用于分析单个酶切位点或短的DNA序列[7-8]。随着高通量测序技术的发展及人们对染色质可及性区域了解的增强,现已衍生出许多鉴定染色质可及性的研究方法,主要包括脱氧核糖核酸酶Ⅰ超敏感位点测序(deoxyribonuclease Ⅰ hypersensitive site sequencing, DNase-seq)[9]、甲醛辅助性调控元件分离测序(formaldehyde-assisted isolation of regulatory elements followed by sequencing, FAIRE-seq)[10]、微球菌核酸酶辅助分离核小体测序 (micrococcal nuclease-assisted isolation of nucleosomes sequencing, MNase-seq)[11]、核小体定位和甲基化组测序 (nucleosome occupancy and methylome sequencing, NOMe-seq)[12]以及染色质转座酶可及性测序 (assay for transposase-accessible chromatin with high-throughput sequencing, ATAC-seq)[13]。根据获取染色质可及性区域的方式,可将这些方法分为两类:第1类是直接对染色质可及性区域进行测定,主要有DNase-seq、ATAC-seq和FAIRE-seq;第2类是通过定位核小体,从而间接测定可及性区域,包括MNase-seq和NOMe-seq。DNase-seq建库较为烦琐,且难以检测到与染色质结合时间较短的转录因子[14]。FAIRE-seq方法中甲醛与DNA交联条件难以把握,MNase-seq受酶浓度和切割温度影响较大,NOMe-seq则需要大量的测序读数。这4种技术还有一个共同的局限性,即需要的细胞核数量均巨大(表1)。
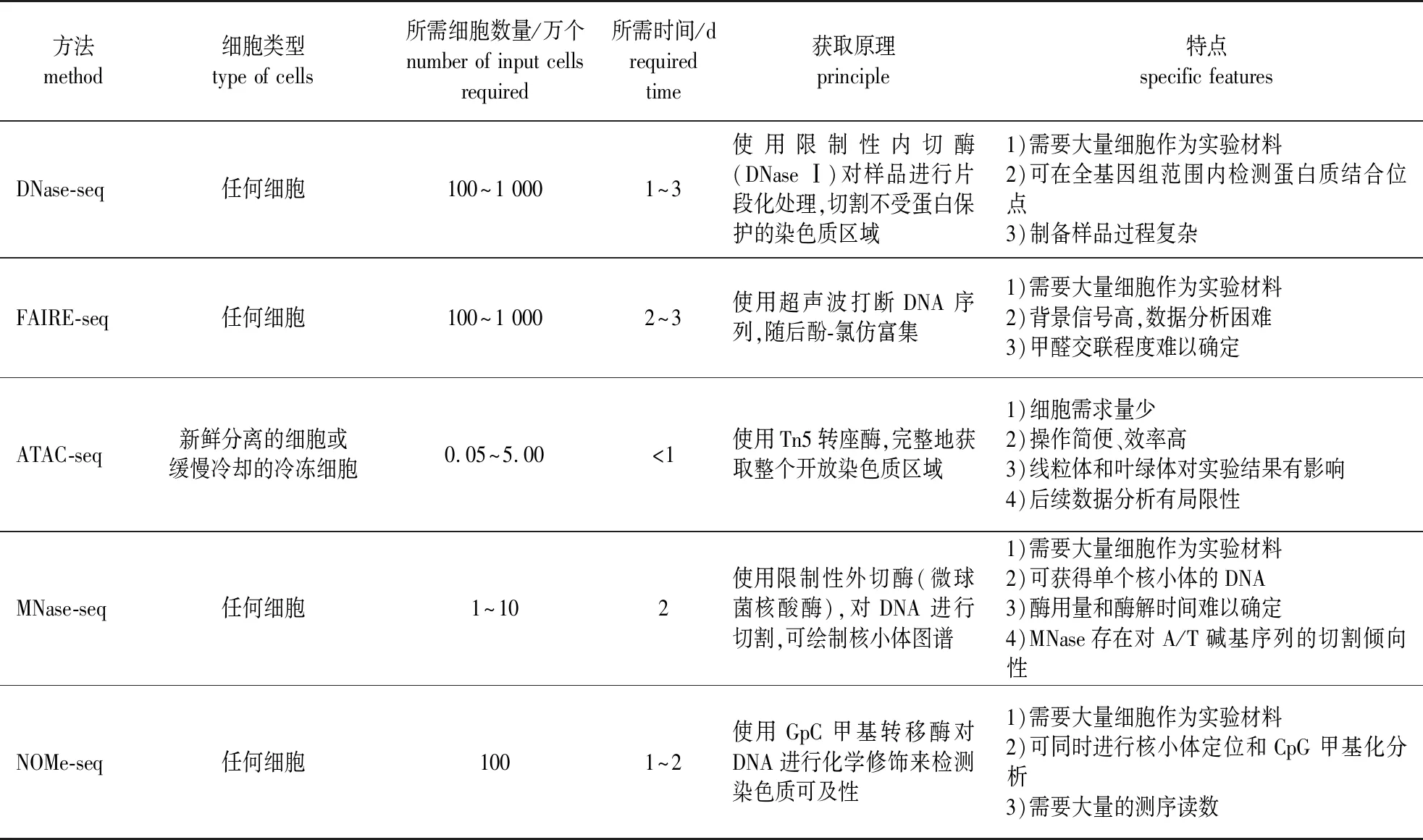
表1 5种研究染色质可及性方法的比较
2013年,Buenrostro 等[13]利用Tn5转座酶开发的ATAC-seq具有所需细胞数量少、建库简便、重复性好等优点。因此,该技术已成为进行真核生物全基因组染色质可及性研究最有效的方法。利用该方法已经在人类和动物中成功构建了全基因组的染色质可及性图谱,为揭示基因表达调控机制、识别转录因子及其同源转录因子结合位点奠定了坚实的基础。然而,与动物细胞相比,植物细胞受到细胞壁和叶绿体的干扰,细胞核不易获得,导致在植物上开展的染色质可及性的研究严重滞后于动物。笔者主要概述了ATAC-seq技术在植物中的应用情况,展望了ATAC-seq在植物中的应用潜力,以推动ATAC-seq在植物表观基因组学研究中的应用。
1 ATAC-seq技术
1.1 ATAC-seq的原理和过程
ATAC-seq利用DNA转座子的原理[15],将带有已知DNA序列标签的Tn5转座酶与DNA进行孵育,在“剪切和黏贴”机制下[16],将裸露的DNA片段在转座酶的作用下置换出来,然后通过特异标签引物构建测序文库来鉴定全基因组ACRs或开放染色质区域(图1)。
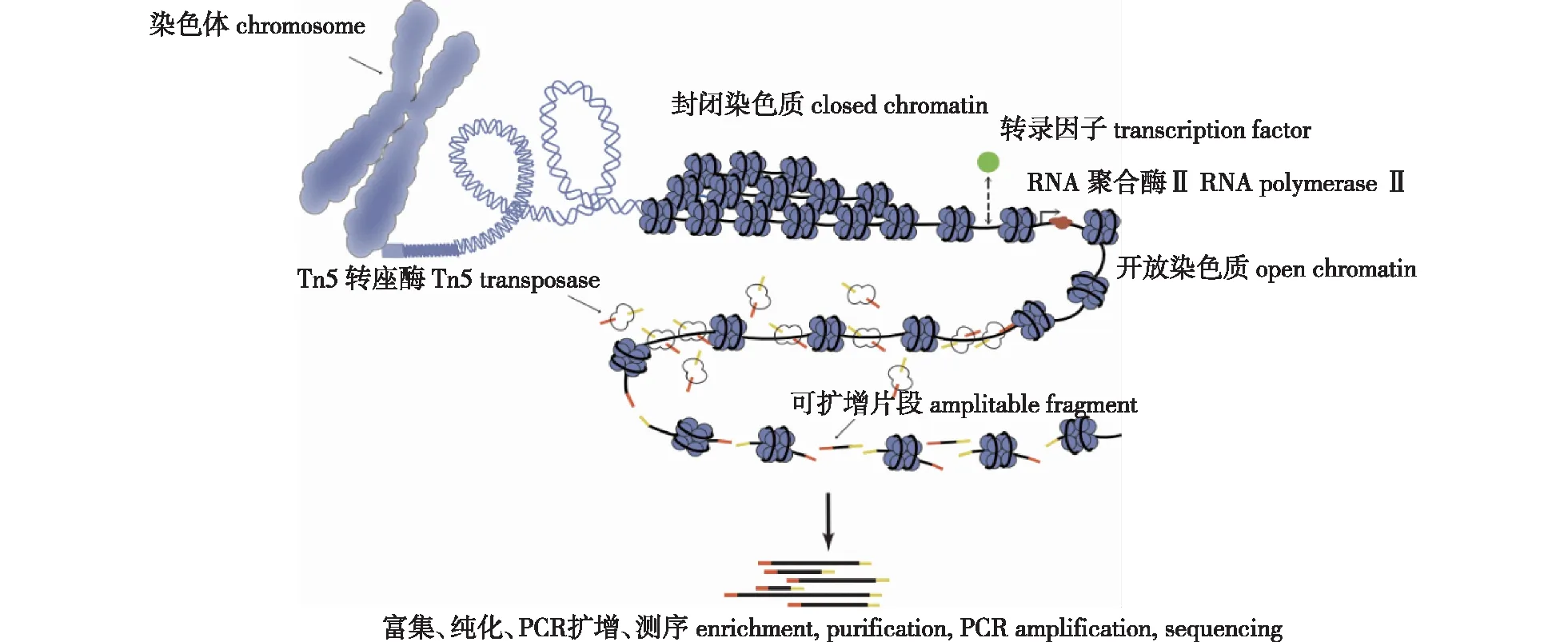
图1 ATAC-seq原理示意图Fig.1 The schematic of ATAC-seq principle
染色质可及ATAC-seq文库的构建主要包括3个步骤:①核制备。可采用液氮研磨法或机械切割法,液氮研磨会导致组织完全破碎,易产生更多的组织碎片[17];若采用切割法,需要将所有的试剂及器皿冷却到4 ℃,以最大限度保证核膜的完整性[18]。②转座并纯化。将提取的细胞核立即重悬于转座酶反应混合物中以产生DNA片段,随后利用试剂盒纯化DNA。③PCR扩增及文库构建。纯化的DNA片段PCR扩增以构建文库,文库扩增所需的最佳循环数,需要用qPCR确定[19],符合要求的文库即可在高通量测序平台上测序[20]。2016年,Lu等[21]首次将ATAC-seq应用到植物中,绘制了拟南芥(Arabidopsisthaliana)根部的染色质图谱。目前该方法已在水稻(Oryzasativa)、番茄(Solanumlycopersicum)、小麦(Triticumaestivum)和高粱(Sorghumbicolor)等植物中成功运用[22-25]。
1.2 ATAC-seq优缺点
与DNase-seq、FAIRE-seq、MNase-seq及NOMe-seq相比,ATAC-seq有3个优点:① 所需的样品量少,500~5 000个细胞就可以建库测序,样品量减少了将近1 000倍;② 实验操作相对简单,2~3 h就可以完成建库工作;③ 结果可信度高,重复性好[26]。ATAC-seq技术也存在局限性,主要包括以下3个方面:首先是细胞器DNA对核基因组的干扰。对于植物来说,分离细胞核时会受到细胞器DNA的干扰,因为Tn5转座酶作用于核外的遗传物质[21, 27-28],从而降低了核基因组的测序读数比例,减少了可用于识别开放染色质区域的信息量。其次,Tn5插入事件及PCR重复对实验的影响[29]。ATAC-seq需采用PCR扩增构建文库,然而无法准确识别相同的片段是由 PCR 扩增所产生,还是由不同的Tn5 插入在相同位置上所产生。若在后续的分析中这些相同的片段被认为是PCR 扩增所引起的重复片段而被去除,将会对染色质可及性区域及转录因子足迹的鉴定造成影响。最后,细胞异质性的限制。细胞是生物体最基本的结构和功能单位,各细胞的基因表达模式是不同的,由于分离单个细胞技术平台的限制,常规测序获得的是群体细胞平均染色质的状态,忽略了细胞的异质性。然而,一些重要生物学过程的发生往往是单个细胞在功能上产生了突变而导致的结果,因此在单细胞层面上研究染色质可及性,对解析生物体发育、分化等基础生物学问题尤为重要。
2 ATAC-seq的优化
ATAC-seq技术虽然摆脱了像其他技术需要大量起始材料及长时间等条件的限制,但仍然有影响其准确性的因素[28],如细胞器DNA、PCR重复及细胞异质性等。为解决这些问题,研究人员从3个方面提出了优化方案。
2.1 植物细胞核提取方法的优化
细胞核是一种高度特化、复杂的细胞器[30],它既是遗传信息库,也是细胞代谢和遗传的控制中心,获得高质量的细胞核是进行ATAC-seq实验的基础。不同细胞核提取方法采用的步骤相似,主要包括组织破碎、过滤、离心、溶解及密度梯度离心[31]。为了降低叶绿体和线粒体DNA对实验的干扰,曲瑞红[32]和Bajic等[19]均采用了蔗糖密度梯度离心来分离细胞核。不同的是前者利用酶解法释放原生质体,再通过梯度离心法收集细胞核。而后者先采用含有非离子表面活性剂的裂解缓冲液裂解细胞器,然后用sucrose sedimentation (crude)来纯化细胞核。Deal等[33]开发了特定细胞类型标记的细胞核分离方法(isolation of nuclei tagged in specific cell types, INTACT),主要包括转基因植物载体构建、生物素连接酶标记和磁珠分离纯化3个步骤。Lu等[21]将荧光激活核分选(fluorescence-activated nuclei sorting, FANS)[34]技术引入到ATAC-seq中,确保了提取所得细胞核的完整性,且制备用于流式细胞术的样品只需组织破碎、过滤、离心3个步骤[35](表2)。

表2 3种纯化细胞核方法的比较
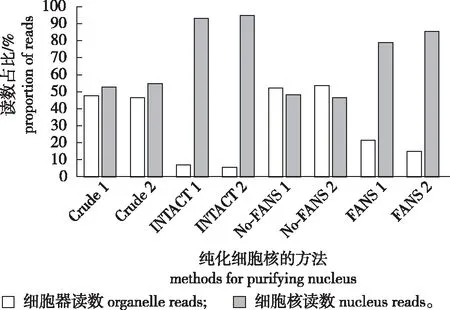
图2 Crude、INTACT和 FANS 纯化的拟南芥根尖细胞核的ATAC-seq 读数[21, 23]Fig.2 ATAC-Seq reads from Crude, INTACT and FANS-Purified arabidopsis root tip nucleus
Sucrose sedimentation、INTACT及FANS 3种制备细胞核的方法,映射到细胞核基因组的读数占比有显著差异。Crude-ATAC-seq得到的结果约有52.52%映射到核基因组,47.48%映射到细胞器基因组;INTACT-ATAC-seq则有92.94%映射到核基因组,7.06%映射到细胞器基因组;FANS-ATAC-seq得到的结果则有78.70%映射到核基因组。因此,与Crude-ATAC-seq相比,FANS-ATAC-seq和INTACT-ATAC-seq得到的测序读数中细胞器基因组污染较少,数据利用率更高(图2),且INTACT-ATAC-seq和FANS-ATAC-seq得到的结果与DNase-seq数据集相比,有很高的重叠率[23, 36]。此外,在拟南芥花序分生组织的侧生器官建成细胞(lateral organ founder cells, LOFCs)中使用FANS-ATAC-seq检测到的转座酶高敏位点(transposase hypersensitive sites, THSs)和DHSs之间存在显著的相关性[37-38],这些实验结果均表明改进后的ATAC-seq可以得到高质量的数据集。
2.2 独特分子标签ATAC-seq技术的应用
Tn5转座酶介导的ATAC-seq能够优先将测序接头插入到“无核小体区域”以实现对染色质可及性区域的捕获。由于在建库过程中需要进行PCR扩增,不同片段的扩增效率存在差异,会产生一些端点相同的重复片段,然而使用常规的ATAC-seq无法从这些重复片段中识别出独立产生的相同Tn5插入片段[29],因此会丢失一些有用的片段从而影响染色质可及性区域及转录因子足迹的鉴定。为了解决上述问题,Zhu等[29]将带有TruSeq测序接头的独特分子标签(unique molecular identifiers, UMIs)引入到标准的实验流程中,建立了UMI-ATAC-seq 技术。该技术在PCR扩增之前给每个片段加上特有的标签,经过扩增测序后,只需把坐标一致并带有相同标签的片段当作PCR重复去掉即可。因此,UMI-ATAC-seq技术通过引入独特分子标签来挽回误认为是PCR重复扩增而被去除的片段,而这些片段对于鉴定转录因子足迹有重要意义,研究发现基于独特分子标签去重的方法虽然只挽救了约6%的读数,但可以多鉴定出约50%的转录因子足迹,且这些足迹具有生物学意义[39]。此外,在构建单细胞转录组文库时,也可以通过对每条转录本添加独特分子标签作为特定标记,进行基因表达定量分析,以解决cDNA扩增过程中PCR偏好问题[40]。与ATAC-seq相比,UMI-ATAC-seq具有成本低、实验操作简单等优势,能够为精准识别染色质可及性区域及转录因子足迹提供科学依据,具有广阔的应用前景。
2.3 单细胞ATAC-seq技术的建立
传统的组学通常是以整个器官或组织为整体进行测序,虽然可以在器官或组织水平上解析许多生物学问题,但掩盖了细胞的异质性[41]。随着生物信息学分析方法及测序技术的迅速发展,衍生出单细胞ATAC-seq(single-cell ATAC-seq, scATAC-seq),研究人员可以在更精细的水平上揭示单个细胞分化过程中的染色质可及性变化及基因转录活跃的部分。该技术目前已广泛应用于生物学和医学领域,并在植物学研究中显示出巨大的潜力。Dorrity等[42]首次将scATAC-seq应用于植物领域并绘制了拟南芥根部的染色质可及性图谱,成功鉴定出拟南芥根部的主要细胞类型。另外,研究者发现在单细胞水平上进行多组学分析对于探究转录调控的潜在机制、建立顺式调控元件与特定基因的调控关系及解释复杂的生命现象等方面扮演重要的角色[39],相关研究表明细胞间异质性的可及性区域与基因转录异质性具有显著相关性[43]。例如联合scATAC-seq和单细胞转录组测序(single-cell RNA-seq, scRNA-seq)可以在单细胞水平上分析出所有活跃的调控序列,从而进一步研究其上游的转录因子。Famer等[44]为揭示染色质可及性对基因表达的影响,整合了scRNA-seq和scATAC-seq的信息,发现拟南芥根组织细胞类型特异性标记基因表现出细胞类型特异性的染色质可及性模式。近年来,研究人员开发了单细胞核染色质可及性和mRNA测序技术(single-nucleus chromatin accessibility and mRNA expression sequencing, SNARE-seq)[45],该技术可以对单个细胞的转录组和染色质可及性进行关联测序。与单独使用scRNA-seq和scATAC-seq相比,该方法能更准确且全面地描述单细胞的类型以及基因表达调控的差异。
3 ATAC-seq在植物领域的应用进展
目前,ATAC-seq技术在植物领域的研究应用主要集中在一些模式植物中,如拟南芥、水稻等,在落花生(Arachishypogaea)、黄花蒿(Artemisiaannua)、高粱及山葡萄(Vitisamurensis)等植物中也开展了少量相关研究。该技术主要应用于染色质可及性图谱绘制、抗逆机制解析、表观修饰鉴定及调控元件识别等方面。
3.1 染色质可及性图谱绘制
染色质结构在确保基因表达的精确调控方面起着关键作用,而染色质可及性在很大程度上决定了下游基因的表达变化,研究开放染色质对于理解基因表达及转录因子调控网络如何在植物中协调以促进分化、生长及发育至关重要。通过绘制染色质可及性图谱可以获得与基因表达相关的染色质特征、转录因子结合位点以及不同样本间图谱的动态变化。Zhou等[46]研究发现,与青蒿素合成途径相关的基因DBR2和CYP71AV1在青蒿腺毛中显示出比在叶中更高的可及性,这为转录因子结合提供了场所,表明ACRs可能参与了相关基因的表达。Zhou等[22]对高粱叶片的ATAC-seq和RNA-seq数据进行综合分析,得出表达水平较高的基因在转录起始位点(transcription start sites, TSSs)处为ACRs,因此推测富集在TSSs上的ACRs有助于基因转录的正调控。类似地,金晶[47]也证明了在水稻中基因表达越活跃染色质开放水平越高这一现象。此外,可以通过分析不同细胞类型之间染色质的可及性来推断转录因子结合的差异,从而阐明细胞分化调控机制。Sijacic等[24]采用INTACT-ATAC-seq分别获得了拟南芥茎顶端分生组织干细胞和叶肉细胞特异的ACRs,构建了两个细胞类型特异性的转录因子调控网络,发现与干细胞相关的转录因子参与了生长素介导的信号转导,而与叶肉细胞相关的转录因子则控制叶片发育,结果表明不同类别的转录因子协同产生细胞类型特异性的转录组,并且在相应的细胞类型中优先表达。Maher等[23]对拟南芥根毛和非根毛细胞特有的THSs进行深入分析,找到了特异的基序,并关联到对应的转录因子,发现根毛细胞中表达量更高的转录因子MYB33和ABI5共同驱动了一个根毛细胞转录调控模式,并且靶向影响根毛细胞命运及应激反应的基因。总之,染色质的开放程度与基因表达调控密切相关,通过多组学的关联分析可以从不同的分析思路获取信息并互相补充验证,有助于揭示基因调控的潜在机制[48]。
3.2 抗逆机制解析
环境条件对植物的生长发育和生态分布有重要影响,植物已进化出多级调控机制来调控基因的表达,以提高其对低温、干旱及病虫害等逆境的适应。研究表明,一些转录因子和顺式调控元件不仅可以调控基因的表达,还在抵御生物和非生物胁迫中有重要意义[49]。Ren等[50]利用ATAC-seq绘制了冷胁迫下葡萄的染色质可及性图谱,共鉴定出 CBF4、RAV1等9个TFs,这些转录因子采用不同的途径对冷应激做出调控,同时还发现VaRAV1基因的过表达可以提高葡萄的耐寒性,为阐明冷响应过程中的信号转导提供了理论参考。Wang等[51]通过结合ATAC-seq、RNA-seq和 Ribo-seq分析了茶(Camelliasinensis)在低温胁迫下的染色质可及性、转录和翻译图谱,以研究染色质水平和翻译水平上的调控机制。结果表明,茶在面临低温胁迫时转录和翻译起协同作用,两者之间的皮尔逊相关系数增加到0.68;其次,在低温条件下,茶的染色质可及性图谱发生了明显的变化,大部分THSs都位于远端基因间区域,这表明远程转录调控可能在茶对低温胁迫的响应中发挥了重要作用。Sullivan等[36]利用染色质可及性变化和转录因子足迹鉴定建立了拟南芥在响应高温胁迫时的动态调控网络,Wilkins等[52]、Lockhart[53]则更进一步将染色质可及性与共表达数据和网络推理算法结合,揭示干旱和高温胁迫下水稻的环境与基因调控互作网络(environmental gene regulatory influence networks, EGRINs),获得了更丰富的信息。Ding等[54]采用ATAC-seq和RNA-seq在染色质可及性和转录调控水平上研究了模式受体激活的免疫反应(PAMP-triggered immunity, PTI)和效应蛋白激活的免疫反应(effector-triggered immunity, ETI)之间的关系,解析了两种免疫系统共激活转录变化的机制,发现两种免疫系统联合比PTI能更有效提高防御基因的表达,这与防御基因位点附近的染色质可及性增强相关,该研究更好地解析了植物免疫激活过程中染色质动态变化对基因表达调控的影响。
3.3 表观修饰鉴定
随着植物表观组学研究的不断深入,研究人员已对植物基因组染色质可及性和表观遗传修饰进行了探究,发现植物染色质可及性区域和表观遗传修饰之间的动态相互作用在一定程度上影响染色质可及性,从而在转录或翻译水平上调控基因表达。为了研究染色质可及性区域相关的表观组学特征,通过将ATAC-seq数据与染色质免疫共沉淀测序(chromatin immunoprecipitation followed by sequencing, ChIP-seq)数据进行整合,可以鉴定不同组蛋白修饰的ACRs,不同组蛋白或同一组蛋白的不同氨基酸残基上的修饰决定染色质处于活性或非活性状态[55]。例如:H3K27me3作为一种组蛋白甲基化修饰,是调控ACRs下游基因表达的一个主要的非活性表观遗传标记,使下游基因沉默;而H3K4me3、H3K36me3和H3K56ac等组蛋白修饰是植物中保守的活性标记,位于活性基因转录起始位点且在调控基因表达方面更倾向于与ACRs呈正相关。其次,植物的染色质可及性区域虽然缺乏5-甲基胞嘧啶(5-methylcytosine, 5mC)修饰,但染色质可及性区域为相关表观遗传调控因子的招募提供了空间,从而在ACRs相关区域及其周围进行表观遗传标记,以促进相关基因的DNA甲基化,研究表明,基因内部具有ACRs 时,该基因的CG甲基化水平高于无ACRs的基因[22]。同时,DNA甲基腺嘌呤 (N6-methyldeoxyadenosine, 6mA)作为一种非典型的DNA甲基化修饰,在植物基因组中发挥着重要的生物学功能。在水稻中,位于启动子中的6mA与基因沉默有关,但在基因内部的6mA与基因活性相关[56],Zhou等[22]进一步证实6mA会与ACRs相互作用以促进基因表达。另有研究表明,参与高温胁迫的关键基因HsfA1和HSP70的表达与6mA呈正相关[57],Liang等[58]发现水稻基因组中的ATAC信号与6mA修饰位点重叠。因此,推测染色质可及性变化和6mA共同调控以应对高温胁迫响应。此外,环境条件会诱导植物染色质结构的改变进而影响调控蛋白与调控元件的结合[59],染色质结构变化与DNA甲基化和组蛋白修饰一样,均可以调节基因的表达。Jégu 等[60]利用ATAC-seq研究表明染色质重塑复合物SWI/SNF的亚基BAF60调控拟南芥下胚轴发育相关基因的表达,在光照条件下,BAF60可以通过影响该基因的染色质可及性,从而抑制了下胚轴伸长。
3.4 顺式调控元件识别
真核生物中基因的表达是通过调控蛋白与启动子、增强子等其他顺式调控元件的相互作用来实现的。其中,增强子(enhancer)是重要的顺式调控元件,能够有效增强基因的转录,其序列和活性的差异在种间和种内的表型变异中起重要作用,但增强子与靶基因的距离和方向不确定,可能位于启动子区域的上游、下游或者基因的内部,因此增强子明显比启动子更难被定位[61-62]。已有大量研究显示,使用增强子捕获和QTL定位技术可以揭示增强子在基因组中的大致位置,但这些技术费时费力[63]。通常情况下,增强子与一些转录因子结合时,会将核小体去除,从而导致局部染色质完全开放,因此可以利用ATAC-seq技术或DNase-seq技术检测染色质可及性区域进而预测增强子,目前基于染色质构象已在拟南芥中开发出了全基因组预测增强子的方法[64]。Huang等[65]利用ATAC-seq技术在大豆(Glycinesoja)中发现远端染色质可及性区域(距离最近基因≥2 kb处)与先前在该物种中报道的Linc RNAs (long intergenic noncoding RNAs)位点重叠[66],而Linc RNAs通常被认为是增强子RNA(enhancer RNAs,eRNAs)[64, 67],这表明远端染色质可及性区域可能是潜在的增强子。Schwope等[68]利用ATAC-seq对葡萄中鉴定出的染色质可及性区域进行了亚硫酸氢盐测序,发现远端染色质可及性区域呈现低甲基化水平,还存在大量转录因子结合位点,这与Calo等[69]提出的增强子往往是低甲基化的观点一致。Lu等[70]通过研究13种被子植物的染色质可及性,表明所有植物中均存在远端染色质可及性区域,其所占比例与基因组大小呈正相关,且观察到保守非编码序列在远端染色质可及性区域中显著富集。另外,Ricci等[71]使用ATAC-seq与多个染色质分析技术相结合,证明了玉米基因组远端ACRs存在大量的顺式调控元件(cis-regulatory elements,CREs),并结合自转录活性调控区测序,结果显示远端调控元件CREs具有增强子活性,调控基因表达。此外,通过比较不同物种及不同发育阶段的ATAC-seq数据,可以更深入地探究调控序列的进化特征,Yan等[72]预测增强子在拟南芥花序发育的不同阶段存在高度的特异性,其序列在十字花科植物中高度保守,Marand等[73]通过比较玉米(Zeamays)和拟南芥根部的发育轨迹,发现两者韧皮部发育过程中的顺式调控模式具有高度的保守性。
4 展 望
ATAC-seq技术自2013 年建立以来,便以材料需求量少、实验操作简便、重复性好等优点成为真核生物研究表观遗传调控的重要技术手段,已在人类[74]及小鼠[75]等模式动物的基因组调控元件鉴定、转录因子结合位点识别及转录调控机制的解析等研究领域发挥了不可替代的作用,展现出广阔的应用前景。相较于动物而言,ATAC-seq技术在植物中的研究应用还处于起步阶段,相关研究主要集中在拟南芥和水稻等模式植物中。随着高通量测序技术的不断进步,以及ATAC-seq技术的进一步完善,该技术必将在植物表观基因组学研究领域发挥重要作用。
获得高质量的细胞核是开展ATAC-seq实验的基础。植物细胞的细胞壁和叶绿体,常常会对细胞核的提取产生影响,使得植物高质量细胞核的制备难度远比动物的大。尽管研究者已对植物细胞核的制备进行了多种优化,出现了Crude、INTACT及FANS[21, 23]等制备细胞核的方法,但这些研究均以拟南芥为实验对象。从笔者提取杨树(Populusspp.)细胞核的经验来看,在拟南芥、水稻、玉米等草本植物中适用的方法,在制备杨树细胞核时还需要进一步优化,才能取得较好的结果。因此,目前主要依据模式植物建立的细胞核制备方法要想成功应用到其他植物尤其是其他木本植物中,还需要根据不同植物材料的特点有针对性地进行优化。
林木世代周期长、遗传杂合度高、目标性状受多基因调控,这增加了揭示林木目标性状遗传机制的难度,从而限制了林木目标性状的遗传改良进程。近年来,ATAC-seq已成功应用于茶和葡萄等木本植物的抗逆机制解析及顺式调控元件识别[51, 68]。可以预见,随着林木功能基因组学研究的深入及高通量测序技术的迅猛发展,ATAC-seq必将在解析木材性状遗传机制这个林木复杂性状研究的热点领域中发挥更重要的作用。
