类似转移性乳腺癌的Rosai-Dorfman病1例
2022-10-15郭圆圆郑占才王清扬
郭圆圆,郑占才,王清扬,佟 杰*
(1.中日友好医院病理科,北京 100029;2. 中日友好医院皮肤科,北京 100029)
患者女性,66 岁,因右腹部及右下颌皮肤肿物两年余就诊。2年前,患者右腹部及右下颌相继出现硬币大小红斑,伴轻度压痛,无搔痒及脱屑;后红斑逐渐扩大隆起呈斑块状,为明确诊治,来我院皮肤科就诊。患者5年前因乳腺癌行根治术后,口服阿那曲唑至今;因甲亢长期服用甲硫咪唑,甲功控制可。发病以来无发热、乏力、体重减轻等。体检:全身浅表淋巴结未及肿大,各系统检查未见异常。皮肤科检查:右腹部及右下颌暗红色、圆形、不规则增生性斑块隆起,右下颌部斑块3.8cm×4.4cm,右腹部斑块4cm×5cm,无鳞屑、破溃及出血,中等硬度,触之有弹性,压痛阳性(图1,见封底)。实验室检查:B 超示右颈部单发淋巴结肿大,1.6cm×0.4cm;右侧腋窝单发淋巴结肿大,最大径0.7cm。余实验室检查未见异常。根据患者皮损特点、乳腺癌病史及淋巴结肿大情况,拟诊断转移性乳腺癌收入院。
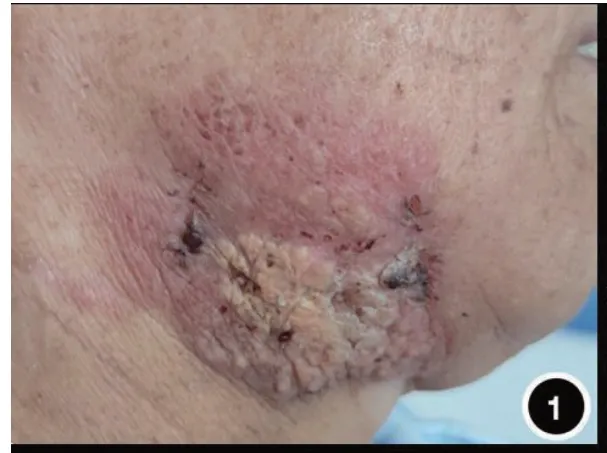
图1 患者右下颌皮损。
病理检查:右下颌皮损活检示病变主要位于真皮,可见大量浆细胞、淋巴细胞及组织细胞带状浸润,部分组织细胞胞浆内可见多个形态完整的淋巴细胞(图2,见封底)。免疫组化:组织细胞CD68(+),CD1a(-),S100(+)(图3 见封底)。病理诊断:(右下颌)结合病理和免疫组化结果,考虑Rosai-Dorfman 病(RDD)。后行右腋下淋巴结穿刺活检示淋巴结结构尚存,窦区扩张,低倍镜下与皮损病理相似,呈“明暗相间”的组织学特点,暗区主要为增生的淋巴细胞及浆细胞,明区富于组织细胞,部分组织细胞表达S100,并见“伸入现象”;局灶见CD1a阳性的组织细胞增生,伴色素沉着;病理学结果符合RDD 合并皮病性淋巴结炎。腹部皮损完全切除后病理形态及免疫组化均符合RDD,结合患者下颌部皮损及淋巴结穿刺结果,整合诊断为混合型RDD。
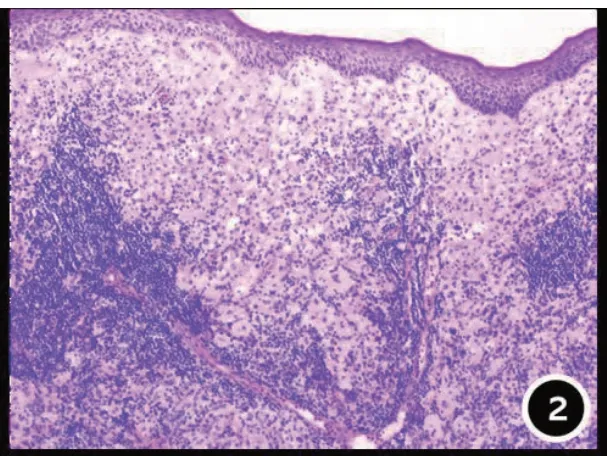
图2 皮损组织病理(HE×100),病变主要位于真皮,可见大量浆细胞、淋巴细胞及组织细胞带状浸润,部分组织细胞胞浆内可见多个形态完整的淋巴细胞。
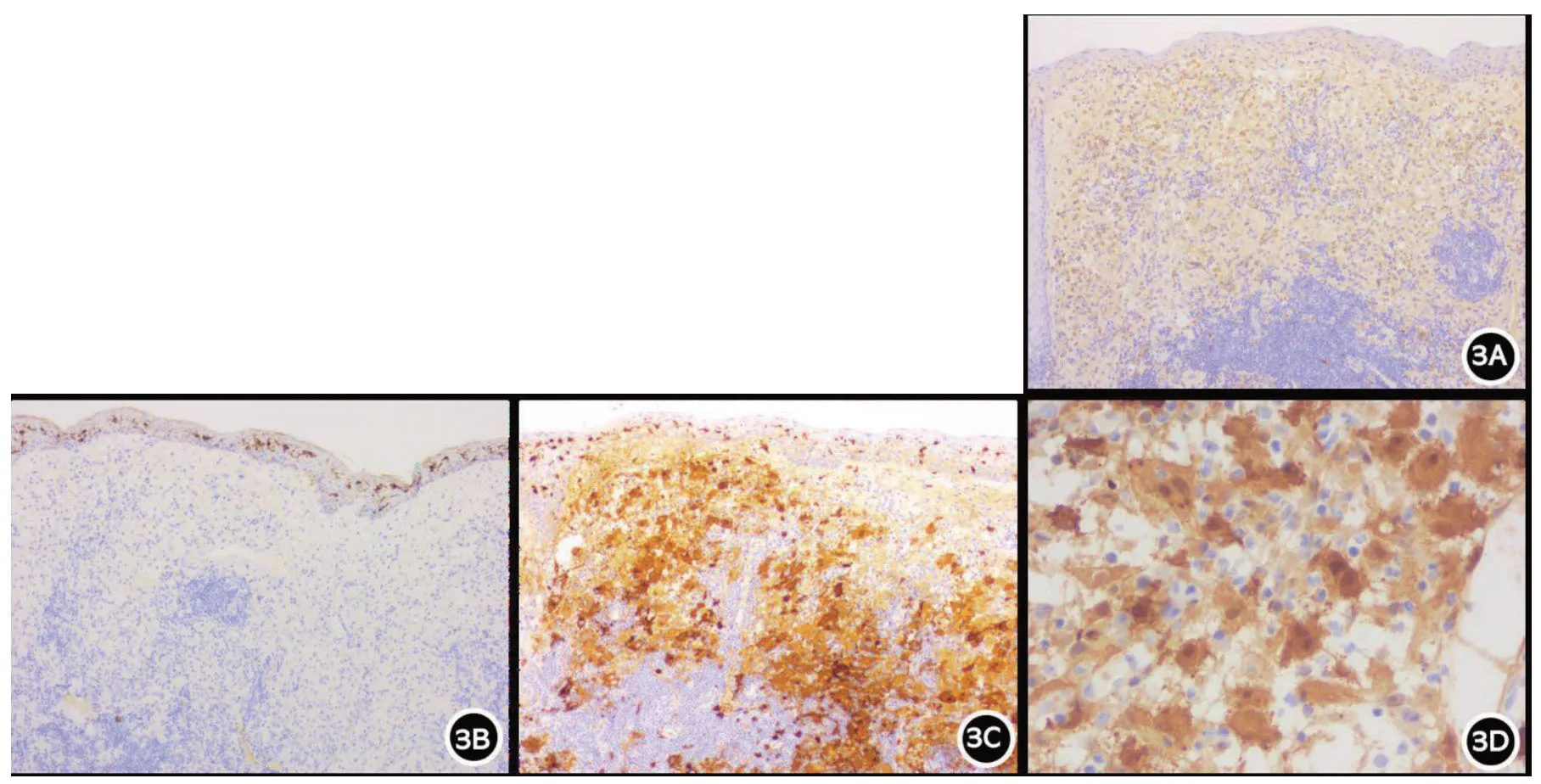
图3 免疫组化检查(En Vision 法;3A~3C:×100,3D:×400),3A:组织细胞CD68(-);3B:组织细胞CD1a(-);3C及3D:组织细胞S100(+)。
治疗:考虑患者下颌部皮损较大,行手术治疗的难度及对美观的影响,仅局部每月给药1 次:复方倍他米松注射液1ml+利多卡因2ml,局部封闭,并随诊淋巴结增大情况。现淋巴结未触及肿大;腹部未见复发;下颌皮损好转变薄。
讨论 Rosai-Dorfman 病(RDD)又称窦组织细胞增生症伴巨大淋巴结病(sinus histiocytosis with massive lymphadenopathy,SHML),被认为是一种少见的良性组织细胞增生性病变,主要表现为双侧颈部淋巴结对称性、无痛性肿大,部分患者伴有发热、体重降低、血沉增快、多克隆性丙种球蛋白血症等。根据其病变累及范围,临床上常将该病分为3 种亚型:①淋巴结型:病变仅位于淋巴结;②结外型:仅有淋巴结外病变而不伴淋巴结病,以皮肤、软组织、眼眶、鼻窦、骨等处较为好发[1];③混合型。其中仅有皮肤损害的原发性皮肤Rosai-Dorfman 病(CRDD)较为罕见,仅占RDD 患者的3%[2],皮损好发于面部,主要表现为黄色或红褐色丘疹、结节或浸润性斑块,可单发或多发,亦可表现为环状肉芽肿样、脂膜炎样、脓疱样或痤疮样等,部分可破溃,多无明显自觉症状。与常发生于中青年白色人种男性的RDD 不同,74.5%的CRDD 患者为亚洲人,并以中老年女性最为常见,预后较好。因CRDD 独特的人口学特征,发生部位及预后,2016年将其从RDD中单独分出[3]。
本病的病因学和发病机制仍不清楚,分子遗传学和分子生物学检测未发现特征性肿瘤单克隆增生表现,推测仍属反应性病变。有研究认为,RDD是一种未确定类型的免疫缺陷性疾病,可以促进血液中单核细胞进入淋巴结内或结外组织[4]。正常情况下,随着单核细胞的分化成熟,其髓相关蛋白(myeloid-related protein,MRP)8 和14 表达会逐渐丢失,而RDD 的组织细胞仍表达MRP8 及MRP14,因此认为RDD 的组织细胞可能是具有分化成熟“缺陷”的单核细胞,这些“缺陷”可能导致了RDD 的组织细胞仅具有吞入而缺乏分解的功能(即镜下的“伸入”而非“吞噬”现象)[5]。此外,亦有报道认为其与病原体感染、免疫功能紊乱及IgG4相关性疾病有关[6]。
RDD的临床表现不具有特征性,诊断主要依据其组织病理学改变及免疫组化特点:①低倍镜下呈浅染区和深染区交替形成的弥漫性或结节性病变;②高倍镜下浅染区组织细胞中存在“伸入现象”,吞噬完整的淋巴细胞和浆细胞,部分细胞中可见吞噬中性粒细胞和红细胞;③组织细胞CD68和S100阳性表达,CD1a不表达。该病需与多种疾病相鉴别[7],如朗格汉斯组织细胞增生症(Langerhans cell histiocytosis,LCH)中组织细胞除表达CD68 和S100 外,还表达CD1a,且无“伸入现象”。此外,常伴有大量嗜酸性粒细胞浸润;恶性组织细胞增生症中也可见到组织细胞吞噬现象,但吞噬细胞数目少,吞噬物主要为核碎片、核尘及红细胞,而不是完整的淋巴细胞、中性粒细胞等炎症细胞,并常见核异形及核分裂象。
对单发、较小的皮损,或累及内脏器官及造成局部压迫的RDD 病例,首选手术切除,有报道采用局部注射治疗、光动力治疗等取得了一定的疗效[8,9]。对于病变泛发或行手术或局部治疗后复发的患者,可采用糖皮质激素、免疫抑制剂、生物制剂等系统治疗方法[10]。RDD 目前尚缺乏规范化的治疗方案,治疗方法的选择大多源于经验或参考以往的病例报道,而缺乏大样本多中心的临床对照研究。RDD 通常不威胁患者生命,部分患者可自发缓解,对临床上病变局限且无重要脏器受累或产生压迫症状的患者可选择随访观察,避免过度治疗。
本例患者的诊疗过程有两个特殊之处值得关注:①患者曾有乳腺癌病史,此次出现浸润性皮损及淋巴结肿大,临床表现与转移癌难以鉴别,因此病理学检查区分转移癌及RDD 至关重要。目前有个别报道与本例患者出现类似的模拟癌症转移的皮损及淋巴结肿大[11],RDD 与癌症的关系尚需进一步研究证实。②患者为老年女性,因皮损就诊,浅表淋巴结未触及肿大,仅根据其皮损情况及人口学特征极易诊断为CRDD,因患者乳腺癌病史行淋巴结B 超时发现肿大的淋巴结,后病理证实皮损及淋巴结均为RDD,这就提示我们诊断CRDD 时应进行全面的系统检查排除混合型RDD,避免因漏诊延误患者治疗。
既往报道显示部分RDD 病例存在RAS、PIK3CA、TNFRSF6、MAP2K1、BRAF、ARAF、mTOR、KMT2D 及NOTCH1 等基因改变[12]。本例患者行全基因组外显子测序,发现FGFR3 基因的c.1949A>C p.K650T 错义突变,FGFR3(III型成纤维细胞生长因子受体)隶属FGFR 酪氨酸激酶家族,可与成纤维细胞生长因子结合,并启动下游的MAP、JAK/STAT 和PI3K/AKT 信号通路,最终影响细胞的增殖、生长、迁移与分化。FGFR3 突变在多种肿瘤中均有报道,包括乳腺癌、卵巢癌,尿路上皮癌等[13~15],在RDD 患者中为首次报道,有一定的深入研究价值。
