2-吲哚亚胺构建吲哚并环骨架研究进展
2022-10-14刘万聪马恩典田远航辛子杰李俊龙
刘万聪,张 斌,马恩典,田远航,辛子杰,陈 林,李俊龙,2
(1.成都大学 药学院(四川抗菌素工业研究所)抗生素研究与再评价四川重点实验室,四川 成都 610052;2.成都大学 高等研究院手性药物创新研究中心,四川 成都 610106)
0 引 言
吲哚衍生物在自然界中普遍存在,其具有独特且广泛的生理药理活性[1],具体表现为抗病毒[2]、抗菌[3]、抗癌[4]、抗炎[5]与抗抑郁[6]等.因此,有机化学家和药物化学家们将深入探究含有吲哚并环骨架的合成砌块,开展高效构建吲哚并环骨架等作为重要的研究方向之一[7].近年来,2-吲哚亚胺因易于制备且具有良好的反应活性,常常作为合成砌块被用于构建含吲哚并环的骨架结构.2-吲哚亚胺能够在布朗斯特碱的作用下去质子化,转化形成烯胺负离子,进而对双亲电试剂进行第一次进攻;经由分子内的质子转移后又发生第二次进攻,完成环化过程,实现多样化的吲哚并环骨架的构建(见图1).基于此,本文综述了2-吲哚亚胺参与的(2+2) 环化反应、(3+3) 环化反应与多组分串联环化过程在吲哚并环骨架中的合成应用,以期为吲哚并环化合物合成方法的扩展提供必要的参考.
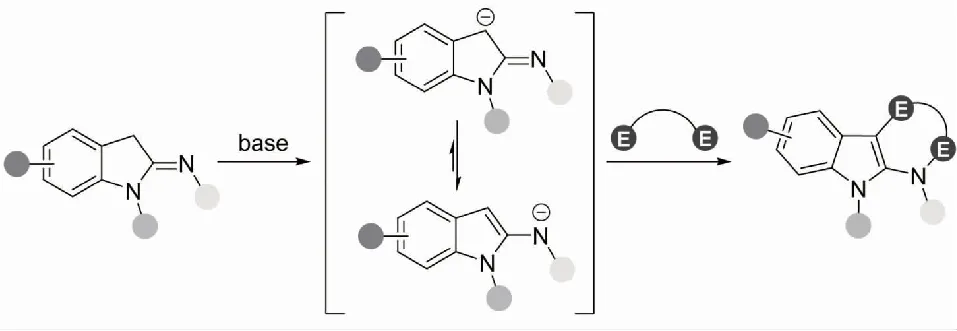
图1 2-吲哚亚胺参与的环化反应过程
1 (2+2) 环化反应合成吲哚并环衍生物
在多样化的环化反应中,(2+2) 环化反应[8]普遍要求光照、Lewis酸或者过渡金属作用等相对较为苛刻的反应条件.所得的四元环化产物也常常在自身较大的环张力下而开环转化为其他环系骨架.但从另一方面思考,扩环过程破坏了四元环状骨架的构建,但是由(2+2) 环化反应介导的扩环过程却成为了部分其他环化反应的有效路径.

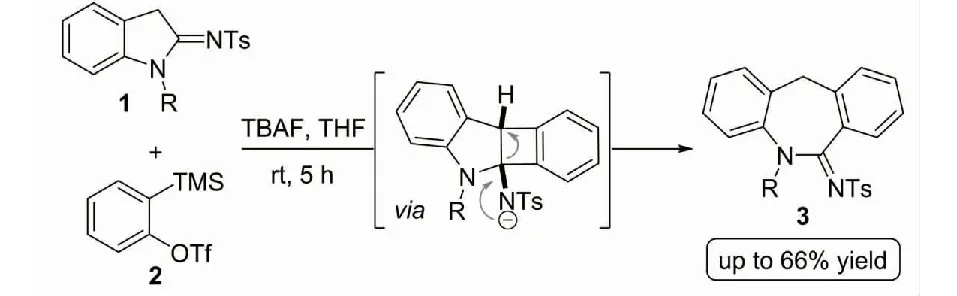
图2 (2+2) 环加成反应介导的二苯并氮杂亚胺衍生物的合成
2 (3+3)环化反应合成吲哚并环衍生物
(3+3) 环化反应合成的六元环状骨架与(2+2) 环化反应合成的不稳定的四元环系骨架相反,其合成的六元环状骨架属于热力学稳定的环系骨架.基于这样的稳定性,在2-吲哚亚胺参与的串联环化方法学研究中,尝试在Michael加成介导的关环反应(michael-initiated ring-closing reaction,MIRC反应)[10]的过程中实现手性控制或发展多样化的双亲电试剂,是围绕2-吲哚亚胺反应开发的两个重要发展方向.
2.1 氮杂环卡宾(NHC)介导的(3+3) 环化反应
2017年,Yi等[11]用NHC催化,以良好到优秀的收率合成了一系列吲哚并哌啶酮衍生物6(见图3),实现了2-吲哚亚胺1和α-溴代不饱和醛4的(3+3) 环化反应.当使用α-溴代肉桂醛(4,R2= Ph,R3= H)作为亲电试剂时,合成的化合物能够在钯—碳的催化下,转化为α-咔啉酮,丰富了该方法的合成价值.

图3 NHC催化的2-吲哚亚胺与α-溴代不饱和醛的(3+3) 环化反应
根据实验结果和已有相关文献报道[12-13],该课题组提出了该(3+3) 环化过程可能的催化循环反应机理(见图4).NHC前体Cat.5先被碱活化,形成活性NHCCat.5A,并与α-溴代不饱和醛4亲核加成,消除卤素生成α,β-不饱和酰基唑鎓阳离子4A;随后,2-吲哚亚胺1在碱的去质子化作用下生成的烯胺负离子与4A加成得到两性离子中间体4B,然后经质子转移形成烯胺负离子4C;最终,内酰胺化得到(3+3) 环化产物6,同时NHC催化剂离去,完成催化循环.
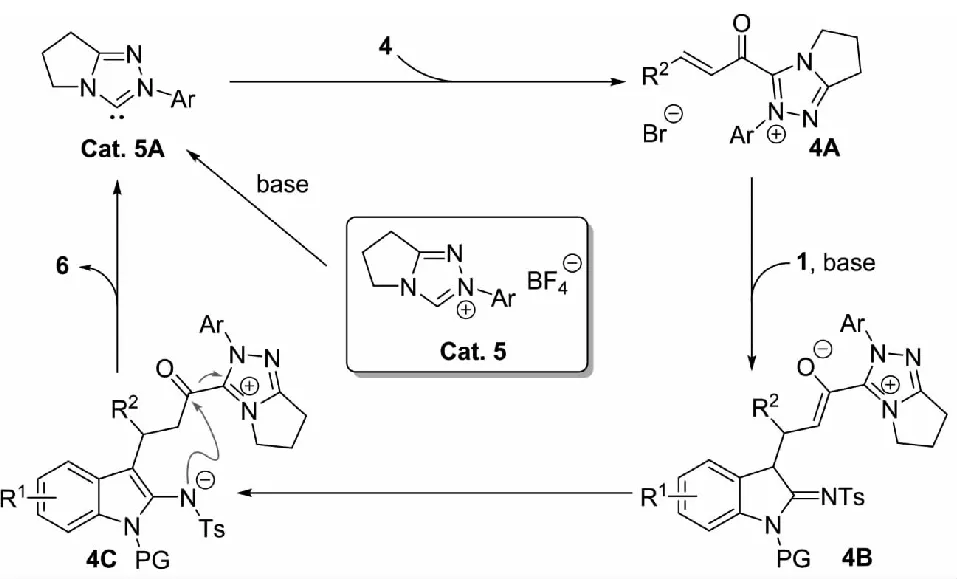
图4 NHC催化的2-吲哚亚胺与α-溴代不饱和醛的(3+3) 环化反应机理
2021年,Ma等[14]通过底物设计及手性催化剂的切换,进一步实现了NHC介导的不对称催化转化.通过NHC催化邻位取代的芳基丙炔酸对硝基苯酚酯7与2-吲哚亚胺类化合物1的不对称(3+3) 环化反应,以中等到优秀的收率及立体选择性,首次实现了α-咔啉轴手性骨架的构建(见图5上部分).并通过密度泛函(DFT)理论计算,阐明了氢键等弱相互作用为控制对映体选择性来源的关键因素.

图5 NHC催化的4-芳基-α-咔啉轴手性衍生物的合成
首先,手性NHC前体Cat.8被碱活化,产生手性NHC,进攻芳基丙炔酸酯7,得到关键的炔基酰基唑鎓中间体7A;随后,在碱的作用下,2-吲哚亚胺1去质子化产生亚胺负离子与7A共轭加成生成丙二烯醇化物中间体7B,该中间体经质子转移和烯醇互变后形成中间体7C,其氮负离子通过分子内亲核进攻羰基碳得到环化产物9的同时NHC离去,完成催化循环;接下来,环化产物9芳构化,对甲苯磺酰基(Ts)从氮转移到氧,形成更稳定的产物4-芳基-α-咔啉轴手性衍生物10(见图5下部分).
2.2 异硫脲介导的(3+3) 环化反应
2019年,Liu等[15]采用手性异硫脲催化剂Cat.12活化β-多氟烷基-α,β-不饱和羧酸酯11,形成双亲电试剂与2-吲哚亚胺1完成了不对称(3+3) 环化反应,以中等到优秀的收率及立体选择性得到了手性吲哚并二氢吡啶酮衍生物6(见图6).但在反应的普适性考察中发现,当把α,β-不饱和羧酸酯11中的β-多氟烷基更换为甲基与丙基等给电子基团时,反应收率下降,这表明该反应明显依赖Michael受体较高的缺电子程度.
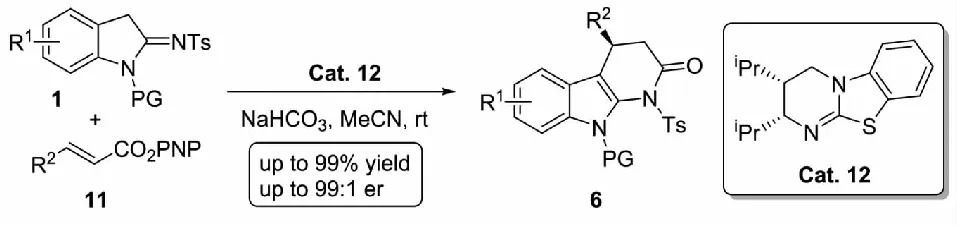
图6 异硫脲介导的α-咔啉酮的不对称合成
根据实验结果及已有的异硫脲催化领域中的研究[16],该课题组提出了可能的催化循环机理(见图7).α,β-不饱和羧酸酯11被手性异硫脲催化剂Cat.12酰基化,生成α,β-不饱和酰基异硫脲离子对11A;2-吲哚亚胺1在碱的作用下被拔质子转化产生的烯胺负离子,与α,β-不饱和酰基异硫脲离子对11A加成,得到烯醇异硫脲鎓11B,经过分子内的质子转移过程产生两性离子中间体11C;最后,氮负离子亲核进攻羰基碳得到内酰胺化的产物6,同时手性异硫脲催化剂Cat.12离去,完成催化循环.而α,β-不饱和酰基异硫脲离子对11A采用s-顺式构象可以解释反应的立体化学结果.
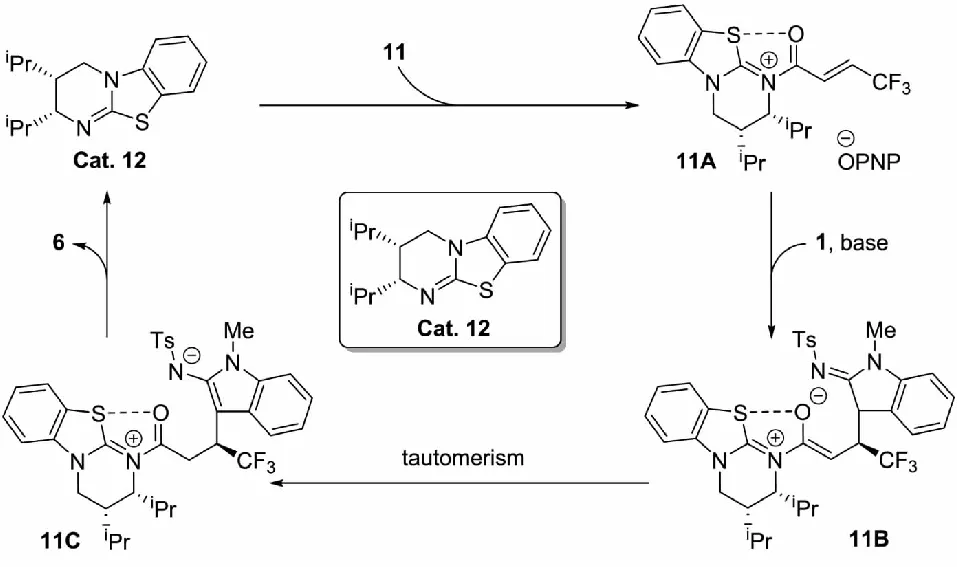
图7 异硫脲介导的α-咔啉酮的不对称合成的反应机理
2.3 叔膦介导的(3+3) 环化反应
叔膦作为一种重要的路易斯碱,常常被用于活化联烯酸酯产生多反应位点的活性中间体,进而参与多种类型的串联环化过程.2021年,Debnath等[17]使用三苯基膦作为催化剂,活化δ-乙酰氧基联烯酸酯13与2-吲哚亚胺类化合物1发生(3+3) 环化反应,并能够通过条件的控制,多样性地得到含有不同饱和度的α-咔啉衍生物(见图8).在室温下,δ-乙酰氧基联烯酸酯13与2-吲哚亚胺1能够反应得到二氢-α-咔啉骨架14.当向反应体系中加入碳酸铯并且升高温度至80 °C时,则能够通过对甲苯磺酰基的迁移带来的芳构化的动力得到α-咔啉衍生物10.尽管这一过程的反应收率尚存在一定的提升空间,但这一工作为α-咔啉衍生物的多样性制备提供了重要的参考.
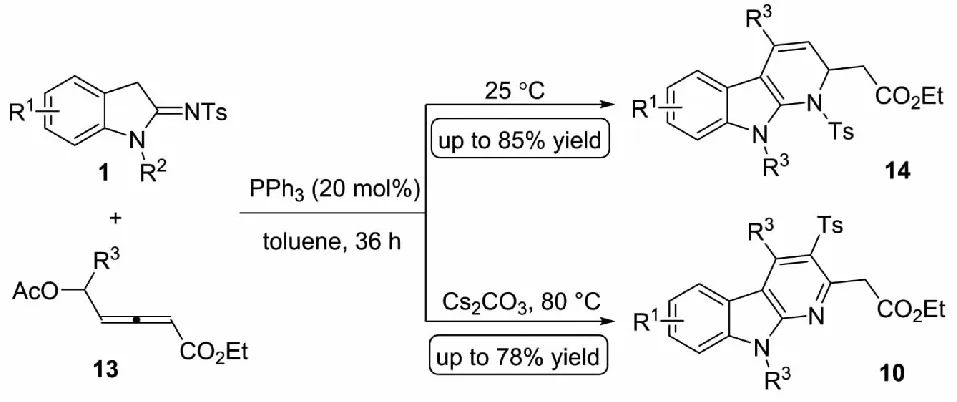
图8 三苯基膦介导的α-咔啉的多样性合成
2.4 叔胺介导的(3+3) 环化反应
在2-吲哚亚胺参与的(3+3) 环化反应中,叔胺主要是作为布朗斯特碱用于完成2-吲哚亚胺的去质子化,进而推动反应的发生.2020年,Chen等[18]在金鸡纳碱衍生的叔胺方酰胺双功能催化剂Cat.16的作用下,α,β-不饱和酰亚胺15与2-吲哚亚胺1发生(3+3) 串联环化反应,以中等到优秀的收率及立体选择性构建了α-咔啉酮手性骨架结构(见图9).在该反应中,奎尼丁胺衍生的方酰胺催化剂Cat.16既作为布朗斯特碱用以活化2-吲哚亚胺1,又作为氢键供体提升α,β-不饱和酰亚胺15的亲核性,通过对底物的双活化模式在促进反应高效进行的同时达到控制产物立体构型的目的.

图9 叔胺方酰胺双功能催化剂介导的α-咔啉酮的不对称合成
合成化学家对硝基烯烃的MBH(morita-baylis-hillman)仲醇衍生的乙酸酯独特的1,2-或1,3-双亲电活性较为关注.2021年,Sivanandan等[19]用三乙烯二胺(DABCO)拔掉2-吲哚亚胺3位的质子,随后转化形成的烯胺负离子亲核进攻硝基烯烃衍生的MBH乙酸酯17,以优秀的收率,区域选择性地得到了一系列α-咔啉衍生物10(见图10上部分).在该反应中,2-吲哚亚胺1在碱的作用下产生的烯胺负离子1A进攻硝基烯烃衍生的MBH乙酸酯17,醋酸根经过SN2的历程离去,生成中间体17A.在碱的作用下,由于硝基的吸电子能力强于酯基,中间体17A对硝基的β位发生分子内Michael加成,形成四氢-α-咔啉衍生物18.随后,四氢-α-咔啉衍生物18在碱的作用下消除1分子亚硝酸和1分子对甲苯亚磺酸,最终生成α-咔啉衍生物10(见图10下部分).

图10 硝基烯烃衍生的MBH乙酸酯与2-吲哚亚胺参与的(3+3) 环化反应
随后,该课题组在此工作基础上,将硝基烯烃衍生的MBH乙酸酯17更换为其衍生的MBH溴化物19,并把反应的溶剂更换为极性较小的甲苯,将反应停留在了消除/芳构化的前一步,以优秀的收率及非对映选择性得到了一系列硝基保留的四氢-α-咔啉衍生物18(见图11)[20].
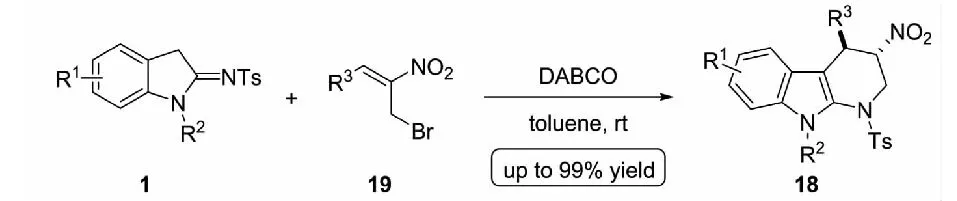
图11 硝基烯烃衍生的MBH溴化物与2-吲哚亚胺参与的(3+3) 环化反应
2022年,He等[21]在手性叔胺催化剂Cat.21的作用下,高效地实现了靛红衍生的MBH碳酸酯20和2-吲哚亚胺1的不对称(3+3) 环化反应,以良好到优秀的收率和高立体选择性产生一系列多官能化的α-咔啉-螺-2-吲哚酮衍生物(见图12上部分).基于实验结果和相关报道,该课题组为这种催化的(3+3) 环化过程提出了一个合理的催化循环(见图12下部分).首先,手性路易斯碱Cat.21加成到靛红衍生的MBH碳酸酯20上,得到亲电铵盐20A;同时,原位生成的叔丁氧负离子作为布朗斯特碱使得2-吲哚亚胺1去质子化,形成亚胺负离子中间体1A;随后,亚胺负离子中间体互变成为烯胺负离子并进攻铵盐20A的γ-位,得到两性离子中间体化合物20B;接下来,催化剂离去,烯胺互变得到烯丙基化产物20D,再经分子内的氮杂-迈克尔加成过程,生成产物22.

图12 手性叔胺催化剂介导的α-咔啉-螺-2-吲哚酮衍生物的合成及机理
2.5 路易斯酸介导的(3+3) 环化反应
在上述反应过程中,2-吲哚亚胺需要预先制备,再作为反应底物参与到环化过程中.2020年,Xu等[22]利用邻氨基苯乙腈23作为2-吲哚亚胺1的前体化合物,在三价铁盐的催化下原位形成2-吲哚亚胺1,进而与1,3-二酮24(见图13上部分)或α,β-不饱和羰基化合物25(见图13下部分)发生(3+3) 串联环化反应,以中等至良好的收率制备了三氟甲基-α-咔啉和酯基取代的α-咔啉衍生物10.
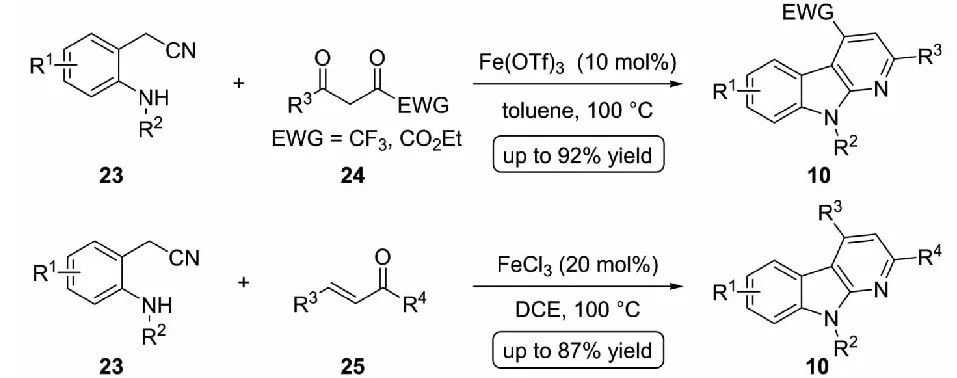
图13 三价铁催化串联环化反应合成α-咔啉衍生物
随后,该课题组提出了该(3+3) 环化过程可能的反应过程(见图14).首先,邻氨基苯乙腈23在铁盐催化下通过分子内环化生成2-吲哚亚胺1,由于2-吲哚亚胺1的C-3位相较于NH2基团具有更强的亲核性,其与24的羰基迅速发生亲核加成反应生成中间体24A;随后,通过分子内缩合与脱水等过程得到目标产物.

图14 三价铁催化串联环化反应合成α-咔啉衍生物的反应机理
次年,基于之前的工作基础,Lu等[23]继续使用三氟甲磺酸铁作为催化剂,活化邻氨基苯乙腈衍生物23,进而与3-二甲胺基烯胺酮类化合物26发生类似的(3+3) 环化反应,高效地选择性合成了一系列2-取代的吡啶并[2,3-b]吲哚衍生物10(见图15上部分).除了与上述过程类似的脱水消除外,在这一工作中,目标产物的生成还伴随了1分子二甲胺的离去(见图15下部分).

图15 三价铁催化邻氨基苯乙腈与3-二甲胺基烯胺酮串联环化反应合成α-咔啉衍生物
3 多组分串联环化反应合成吲哚并环衍生物
在过去的几十年间,多组分反应(multi-component reaction,MCR)的研究探索取得了重大的进展,这一领域的研究成果,为从简单和可变的起始材料构建复杂多样的结构提供了行之有效的合成方法[24-26].多组分化学反应具有操作简单、资源利用率高和高原子经济性等特点,是一类重要的有机化学反应,在新药设计与合成、组合化学和天然产物合成中具有广泛的应用,但来自多组分底物间的竞争反应是其显而易见的挑战.

图16 三组分(3+1+3) 串联环化构建吲哚并氮杂骨架
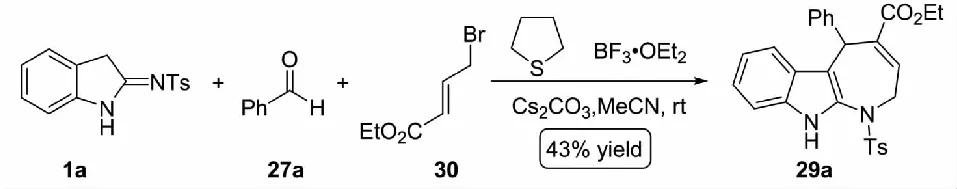
图17 硫醚催化的三组分(3+1+3)串联环化反应
基于多组分反应在构建先导化合物方面所显示出巨大的优势,2020年,Leng等[28]设计了一种三组分(3+2+1) 环化反应来高效合成具有二氢-α-咔啉结构的钩吻吲哚生物碱类似物32(见图18上部分).这一多组分串联合成策略,可以简洁高效地在钩吻吲哚生物碱类似物的核心结构上引入各种取代基和官能团,很好地满足了中药化学对于化合物结构多样化的研究要求.其可能的反应机理(见图18下部分),首先,在碱性条件(DABCO)下,丙二腈31和醛27发生Knoevenagel缩合形成烯基丙二腈33;随后,2-吲哚亚胺1在碱的作用下被拔质子与烯基丙二腈33发生Michael加成,再经过一系列的分子内的质子转移、亲核加成、互变异构与质子化等过程完成环化,得到目标产物.
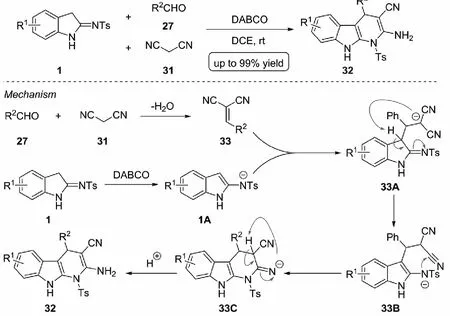
图18 三组分(3+2+1) 串联环化构建二氢-α-咔啉骨架
4 结 语
本文以2-吲哚亚胺参与的串联环化反应作为切入点,详细介绍了其作为含有吲哚环系的合成砌块参与的不同类型的环化反应.尽管目前2-吲哚亚胺被用作合成砌块来构建含有吲哚并环骨架结构的反应实例并不少见.但其参与的串联环化反应中,双亲电试剂的类型还较为局限,基于结构更为多样的新型吲哚并环合成砌块的探索,开发与之反应的新型双亲电试剂的研究仍还处于起步阶段.此外,基于双亲电试剂手性控制的尝试仍然不够充分与深入.因此,尝试在两步串联环化过程中引入手性控制,发展使用结构多样的双亲电试剂,不仅是2-吲哚亚胺参与的串联环化方法学研究中的重要发展方向,也能够为吲哚并环骨架的构建带来新的机遇,具有十分重要的理论意义和现实价值.
