新型含吡啶酮(吡唑)结构的A2a/A2b双靶点腺苷受体拮抗剂的合成及生物活性研究
2022-10-11胡代强付信珍张淑敏
李 志, 胡代强, 付信珍, 张淑敏*, 刘 明*
(1. 滨州医学院 药学院,山东 烟台 264003; 2. 苏州普瑞森生物科技有限公司,江苏 苏州 215004)
肿瘤的免疫治疗在放疗、化疗和靶向药物治疗之后,成为治疗肿瘤疾病的第三种革命性策略[1]。免疫治疗以免疫检查点抑制剂(Immune checkpoint inhibitors, ICI)[2]的临床研究最为成熟,且应用最为广泛[3]。ICI治疗方法给癌症患者带来新的曙光,但是大量数据表明ICI治疗只对部分患者有效,并且9%~29%的患者治疗后会出现肿瘤“超进展”现象[4]。因此,如何激活、增强肿瘤的免疫治疗应答率是肿瘤免疫疗法走向广泛应用的难题之一。
腺苷是肿瘤微环境中免疫应答的重要调节因子[5],其通过作用于腺苷受体(Adenosine Receptor,AR)发挥作用[6-7]。AR是一种G蛋白偶联受体(Protein-Coupled Receptor, GPCR),其在药理学中分为4类:A1、 A2a、 A2b和A3[8-9]。其中,A2aR尤为重要,是被广泛研究的药物靶点之一[10]。腺苷作为一种免疫抑制代谢物,可以与T细胞上表达的A2a受体结合,而这一结合会抑制T细胞的免疫反应,从而使肿瘤细胞产生“免疫逃逸”[11]。A2bR存在于树突状细胞中,在与肿瘤相关的巨噬细胞及骨髓来源的抑制性细胞中高表达[12]。而阻断A2bR可以提高免疫细胞在肿瘤环境中的免疫应答,从而抑制肿瘤生长。A2受体拮抗剂可以与腺苷竞争性地结合A2a或A2b受体,进而保持相关免疫细胞的免疫活性,保留其杀伤肿瘤细胞的能力。目前,进入临床的小分子腺苷受体拮抗剂主要包括:PBF-509[13-14]、 PBF-999、 CPI-444[15]、 CS3005和AB928[16]等。这些药物无论是单用还是与PD(L)-1联用,在肿瘤治疗方面都取得了较好的临床效果。值得注意的是,上述小分子腺苷受体拮抗剂大都只作用于A2aR单靶点,仅有AB928为A2a/A2b双靶点药物。
吡啶酮骨架作为一种兼有N-杂环和酰胺结构单元的化合物,在各种天然产物中都有发现[17-18]。其整合了多种小分子免疫检查点抑制剂药物的设计理念,在保留三唑联嘧啶胺结构基础上,引入了吡啶酮的结构,设计出了一类含有吡啶酮骨架的双靶点A2腺苷受体拮抗剂。再根据生物电子等排原理,将这一类化合物衍生为含有吡唑结构的化合物,合成出了6个结构新颖的双靶点A2腺苷受体拮抗剂(Scheme 1)。所得目标化合物均经1H NMR、13C NMR和HR-MS进行了结构确认,采用cAMP法[22]研究目标化合物对A2aR和A2bR的抑制活性。

Scheme 1
1 实验部分
1.1 仪器与试剂
X-4A型数字显示显微熔点仪;Bruker AVANCE NEO 600 MHZ型核磁共振仪(CDCl3或DMSO-d6为溶剂,TMS为内标);Thermo Q-Exactive型高分辨质谱仪;Waters QDa型液相色谱-质谱联用仪(ESI电离模式);Waters e2695型高效液相色谱仪;Waters ACQUITY型超高效液相色谱仪。
中间体18、20和21按照文献[20]方法合成;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1)3a~3c的合成(以3a为例)
N2保护下,将化合物1(800 mg, 6.5 mmol)、甲基环氧丙烷(2a)(2.34 g, 32.5 mmol)、 Cs2CO3(6.35 g, 19.5 mmol)溶于N,N-二甲基甲酰胺(DMF)(8 mL)中,搅拌下于70 ℃反应12 h。反应结束后,依次用水和乙酸乙酯萃取3次。合并有机相,用饱和NaCl溶液洗涤、无水Na2SO4干燥、抽滤、减压浓缩,残余物经硅胶柱层析(洗脱剂:正己烷/乙酸乙酯=3/1,V/V)纯化得化合物3a780 mg。
分别以环丙基硼酸(2b)、 2-碘代丙烷(2c)为原料,用类似的方法合成化合物3b、3c。
3a: 黄色固体,收率62%; MS(ESI)m/z: Calcd for C10H13NO3{[M+H]+}196.10, found 196.16。
3b: 淡黄色固体,收率61%; MS(ESI)m/z: Calcd for C9H9NO2{[M+H]+}164.07, found 164.12。
3c: 黄色固体,收率61%; MS(ESI)m/z: Calcd for C9H11NO2{[M+H]+}166.09, found 166.21。
(2)4a~4c的合成(以4a为例)
将化合物3a(780 mg, 4 mmol)溶于甲醇(10 mL)中,体系降温至0 ℃,加入NaBH4(228 mg, 6mmol),于25 ℃反应1 h。反应结束后用水和乙酸乙酯萃取3次,合并有机相并减压浓缩除去溶剂,得化合物4a788 mg。用类似的方法合成化合物4b和4c。
4a: 黄色油状液体,收率100%; MS(ESI)m/z: Calcd for C10H15NO3{[M+H]+}198.11, found 198.21。
4b: 黄褐色油状液体,收率97%; MS(ESI)m/z: Calcd for C9H11NO2{[M+H]+}166.09, found 166.24。
4c: 棕色油状液体,收率100%; MS(ESI)m/z: Calcd for C9H13NO2{[M+H]+}168.10, found 166.31。
(3)5a~5c的合成(以5a为例)
N2保护下,将化合物4a(788 mg, 4 mmol)溶于15 mL无水四氢呋喃(THF)中,降温至-5 ℃,依次滴加叠氮磷酸二苯酯(DPPA)(2.2 g, 8 mmol)和1,8-二氮杂二环十一碳-7-烯(DBU)(1.22 g, 8 mmol),在室温下反应10 h。反应结束后用水和乙酸乙酯萃取,合并有机相,用饱和NaCl溶液洗涤,无水Na2SO4干燥,抽滤并减压浓缩除去溶剂,残余物经硅胶柱层析(洗脱剂:正己烷/乙酸乙酯=10/1,V/V)纯化后得化合物5a400 mg。
用类似的方法合成化合物5b和5c。
5a: 黄色油状液体,收率45%; MS(ESI)m/z: Calcd for C10H14N4O2{[M+H]+}223.12, found 223.18。
5b: 淡黄色油状液体,收率51%; MS(ESI)m/z: Calcd for C9H10N4O{[M+H]+}191.09, found 191.21。
5c: 无色油状液体,收率52%; MS(ESI)m/z: Calcd for C9H12N4O{[M+H]+}193.11, found 193.22。
(4)8的合成
N2保护下,依次将化合物6(16.4 g, 0.1 mol)、三异丙基硅基炔(7)(36.5 g, 0.2 mol)、Pd(PPh3)2Cl2(7.0 g, 0.01 mol)、 CuI(1.9 g, 0.01 mol)和三乙胺(30.3 g, 0.3 mol)加入THF溶液(300 mL)中,搅拌下于80 ℃反应过夜。反应结束后用乙酸乙酯和水萃取3次,合并有机相并减压浓缩,粗产品经硅胶柱层析(洗脱剂 ∶二氯甲烷/甲醇=50/1,V/V)纯化得20 g化合物8,淡黄色固体,收率64%; MS(ESI)m/z: Calcd for C15H24ClN3Si{[M+H]+}310.15, found 310.21。
(5)10d~10f的合成(以10d为例)
将化合物8(1.55 g, 5 mmol)、 2-氟苯基硼酸频那醇酯(9d)(1.67 g, 7.5 mmol)、 Pd(dppf)Cl2(350 mg, 0.5 mmol)、 K2CO3(2.07 g, 15 mmol)溶于1,4-二氧六环(20 mL)和水(5 mL)的混合体系中。N2保护下,搅拌下于100 ℃反应4 h。反应结束后用乙酸乙酯和水萃取3次,合并有机相并减压浓缩,粗产品经硅胶柱层析(洗脱剂:正己烷/乙酸乙酯=5/1,V/V)纯化得1.2 g化合物10d。
分别以2-甲基-3-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯甲腈(9e)、 2-甲基苯硼酸频那醇酯(9f)为原料,用类似的方法合成化合物10e、10f。
10d: 淡黄色固体,收率65%; MS(ESI)m/z: Calcd for C21H28FN3Si{[M+H]+}370.21, found 370.14。
10e: 淡黄色固体,收率77%; MS(ESI)m/z: Calcd for C23H30N4Si{[M+H]+}391.23, found 391.17。
10f: 黄色固体,收率82%; MS(ESI)m/z: Calcd for C22H31N3Si{[M+H]+}366.24, found 366.31。
(6)11a~11c的合成(以11a为例)
将化合物5a(370 mg, 1 mmol)、化合物10d(333 mg, 1.5 mmol)、碘化亚铜(25 mg, 0.1 mmol)、四丁基氟化铵(TBAF)(523 mg, 2 mmol)溶于t-BuOH(4 mL)和水(4 mL)中,搅拌下于60 ℃反应过夜。反应结束后用乙酸乙酯和水萃取,合并有机相,减压。粗产品经硅胶柱层析纯化得30 mg化合物11a。
分别以5b、10e和5c、10f为原料,用类似的方法合成化合物11b、11c。
11a: 白色固体,收率51%, m.p.225~226 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.55(s, 1H), 8.02(td,J=7.8 Hz, 1.7 Hz, 1H), 7.67(dd,J=6.8 Hz, 2.1 Hz, 1H), 7.59(d,J=2.3 Hz, 1H), 7.56(m,J=7.8 Hz, 6.9 Hz, 5.1 Hz, 1.9 Hz, 1H), 7.49(dd,J=6.9 Hz, 2.0 Hz, 1H), 7.46~7.20(m, 2H), 6.84(s, 2H), 6.26(t,J=6.8 Hz, 1H), 5.48(s, 2H), 4.77(s, 1H), 3.94(s, 2H), 1.06(s, 6H);13C NMR(151 MHz, DMSO-d6)δ: 163.32, 160.69, 160.64, 160.57, 160.56, 159.03, 157.71, 144.94, 140.29, 138.83, 131.55, 131.49, 129.74, 129.73, 124.79, 124.71, 124.38, 124.20, 124.14, 124.12, 115.93, 115.78, 104.17, 104.11, 103.43, 69.24, 56.34, 49.17, 26.76; HR-MS(ESI)m/z: Calcd for C22H22N7O2F{[M+H]+}436.1892, found 436.1888。
11b: 白色固体,收率49%, m.p.161~162 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.60(s, 1H), 7.89(dd,J=7.7 Hz, 1.4 Hz, 1H), 7.75(dd,J=7.8 Hz, 1.3 Hz, 1H), 7.61(dd,J=7.0 Hz, 2.0 Hz, 1H), 7.55~7.49(m, 1H), 7.46(dd,J=6.9 Hz, 2.0 Hz, 1H), 7.25(s, 1H), 6.91(s, 2H), 6.24(t,J=6.9 Hz, 1H), 5.48(s, 2H), 3.38(tt,J=7.5 Hz, 4.2 Hz, 1H), 2.54(s, 3H), 1.03~0.95(m, 2H), 0.88~0.81(m, 2H);13C NMR(151 MHz, DMSO-d6)δ: 166.03, 163.01, 161.49, 157.62, 144.91, 139.40, 138.46, 138.29, 137.52, 133.00, 132.64, 126.34, 124.62, 124.14, 117.39, 112.58, 104.26, 104.04, 48.98, 31.37, 17.64, 5.76; HR-MS(ESI)m/z: Calcd for C23H20N8O{[M+H]+}425.1833, found 425.1827。
11c: 白色固体,收率54%, m.p.218~219 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.57(s, 1H), 7.81(dd,J=7.0 Hz, 2.0 Hz, 1H), 7.43(dd,J=7.5 Hz, 1.5 Hz, 1H), 7.41(dd,J=6.9 Hz, 1.9 Hz, 1H), 7.35(td,J=7.4 Hz, 1.5 Hz, 1H), 7.33~7.27(m, 2H), 7.23(s, 1H), 6.77(s, 2H), 6.33(t,J=6.9 Hz, 1H), 5.47(s, 2H), 5.09(hept,J=6.8 Hz, 1H), 2.39(s, 3H), 1.29(d,J=6.8 Hz, 6H);13C NMR(151 MHz, DMSO-d6)δ: 167.71, 163.01, 159.62, 157.11, 145.15, 138.05, 137.76, 134.67, 134.42, 130.10, 128.27, 128.21, 125.25, 124.39, 124.30, 104.73, 104.20, 49.13, 45.72, 20.77, 19.43; HR-MS(ESI)m/z: Calcd for C22H23N7O{[M+H]+}402.2037, found 402.2031。
(7)13g和13h的合成(以13g为例)
将化合物7(1.00 g, 7.93 mmol)加入100 ml三口圆底烧瓶中,用15mL DMF溶解,依次加入K2CO3(2.19 g, 15.86 mmol)和1-氯-2-甲基-2-丙醇(2a)(1.12 g, 10.31 mmol),于80 ℃反应17 h。用水和乙酸乙酯萃取并合并有机相,用饱和NaCl溶液萃取,无水Na2SO4干燥,抽滤并减压浓缩除去溶剂,残余物经硅胶柱层析(洗脱剂 ∶正己烷/乙酸乙酯=3/1,V/V)纯化得0.617 g化合物13g。
以环丙基硼酸(2b)为原料,用类似的方法合成13h。
13g: 白色固体,收率39%;1H NMR(400 MHz, CDCl3)δ: 7.58(d,J=2.1 Hz, 1H), 6.97(d,J=2.1 Hz, 1H), 4.28(s, 2H), 1.51(s, 6H); MS(ESI)m/z: Calcd for C9H14N2O3{[M+H]+}199.11, found 199.38。
13h: 类白色固体,收率56%; MS(ESI)m/z: Calcd for C8H10N2O2{[M+H]+}167.08, found 167.28。
(8)14g和14h的合成(以14g为例)
N2保护下,将LiAlH4(115 mg, 3.03 mmol)溶解于10 ml无水THF。降温至0 ℃,将化合物13g(200 mg, 1.01 mmol)的无水THF溶液转移至上述体系,室温搅拌3 h。反应结束后,将体系降温至0 ℃,用2M氢氧化钠溶液0.5 mL淬灭反应。用水和乙酸乙酯萃取3次。合并有机相,用饱和NaCl溶液洗涤,无水Na2SO4干燥、抽滤并减压浓缩除去溶剂得150 mg化合物14g。
以13h为原料,用类似的方法合成14h。
14g: 无色油状液体,MS(ESI)m/z: Calcd for C8H14N2O2{[M+H]+}171.11, found 171.28;直接进行下一步反应。
14h: 淡黄色液体,MS(ESI)m/z: Calcd for C7H10N2O{[M+H]+}139.09, found 139.12;直接进行下一步反应。
(8)15g和15h的合成
分别以14g、14h为原料,类似于化合物5a的合成步骤,得化合物15g、15h。
15g: 淡黄色液体,收率73.3%;MS(ESI)m/z: Calcd for C8H13N5O{[M+H]+}196.12, found 196.28。
15h: 淡黄色液体,收率66%;MS(ESI)m/z: Calcd for C7H9N5{[M+H]+}164.09, found 164.26。
(9)11d的合成
以化合物15g和10e为原料,类似于化合物11a的合成步骤,得化合物11d。
11d: 白色固体,收率10%, m.p.85~86 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.51(s, 1H), 7.81(d,J=0.8 Hz, 1H), 7.54(d,J=0.7 Hz, 1H), 7.43(dd,J=7.5 Hz, 1.5 Hz, 1H), 7.35(td,J=7.4 Hz, 1.5 Hz, 1H), 7.33~7.27(m, 2H), 7.22(s, 1H), 6.73(s, 2H), 5.56(s, 2H), 4.68(s, 1H), 3.99(s, 2H), 2.39(s, 3H), 1.04(s, 6H);13C NMR(151 MHz, DMSO-d6)δ: 167.69, 162.98, 157.10, 145.46, 138.02, 137.40, 134.68, 130.44, 130.10, 128.27, 128.22, 125.25, 123.15, 114.25, 104.27, 68.54, 61.42, 43.46, 26.55, 19.43; HR-MS(ESI)m/z: Calcd for C21H24N8O{[M+H]+}405.2146, found 405.2139。
(10)11e和11f的合成(以11e为例)
25 ℃下,将化合物20(100 mg,0.50 mmol)加入50 mL三口圆底烧瓶中。加入1 mL DMF、 5 mL叔丁醇和3 mL水,搅拌溶解。依次加入化合物15g(106 mg,0.55 mmol)、五合水硫酸铜(2.5 mg, 0.01 mmol)和L-抗坏血酸钠(10 mg, 0.05 mmol),60 ℃搅拌过夜。转移至单口瓶浓缩除去溶剂,硅胶柱层析(洗脱剂:二氯甲烷/甲醇=25/1,V/V)得150 mg化合物11e。
以化合物20和15h为原料,用类似方法合成化合物11f。
11e: 类白色固体,收率41%, m.p.173~174 ℃; 1H NMR(600 MHz, DMSO-d6)δ: 8.51(s, 1H), 7.89(dd,J=7.7 Hz, 1.4 Hz, 1H), 7.75(dd,J=7.7 Hz, 1.4 Hz, 1H), 7.65(d,J=2.3 Hz, 1H), 7.53~7.49(m, 1H), 7.25(s, 1H), 6.87(s, 2H), 6.29(d,J=2.2 Hz, 1H), 5.65(s, 2H), 4.67(s, 1H), 3.99(s, 2H), 2.54(s, 3H), 1.04(s, 6H);13C NMR(151 MHz, DMSO-d6)δ: 167.10, 164.06, 158.64, 146.31, 145.67, 140.45, 139.55, 134.09, 133.72, 133.06, 127.42, 124.77, 118.47, 113.66, 105.42, 104.97, 69.61, 62.42, 47.77, 27.64, 18.71; HR-MS(ESI)m/z: Calcd for C22H23N9O{[M+H]+}430.2098, found 430.2097。
11f: 淡黄色固体,收率53%, m.p.166~167 ℃; 1H NMR(600 MHz, DMSO-d6)δ: 8.53(s, 1H), 7.89(dd,J=7.7 Hz, 1.3 Hz, 1H), 7.78~7.73(m, 2H), 7.51(t,J=7.7 Hz, 1H), 7.25(s, 1H), 6.87(s, 2H), 6.26(d,J=2.3 Hz, 1H), 5.62(s, 2H), 3.70(tt,J=7.4 Hz, 3.9 Hz, 1H), 2.54(s, 3H), 1.02~0.93(m, 4H);13C NMR(151 MHz, DMSO-d6)δ: 167.10, 164.07, 158.62, 146.34, 146.23, 140.45, 139.55, 134.09, 133.72, 131.75, 127.42, 124.91, 118.47, 113.66, 105.44, 105.14, 47.77, 33.14, 18.71, 6.71; HR-MS(ESI)m/z: Calcd for C21H19N9{[M+H]+}398.1836, found 398.1835。
1.3 活性测试
采用cAMP法分别测定合成的化合物对A2a和A2b受体的抑制活性。实验分别使用CHO-K1/ADORA2a/Gα15稳定转染细胞系和CHO-K1/ADORA2b/Gα15稳定转染细胞系[20]。
1.4 分子对接
利用Discovery Studio 2017 R2软件,选取人源A2a腺苷受体A2aR-StaR2-bRIL(PDB ID:5IU4)[21]作为受体模型,A2b腺苷受体的蛋白模型是基于(PDB ID: 6PS7)[22]的同源模型,序列同一性61.92%。利用软件中的CDOCKER模块进行分子对接研究。
2 结果与讨论
2.1 合成
实验过程中,分别对化合物18、13g和11a~11d的实验条件作了进一步优化,结果如表1~3所示。化合物18的合成中,K2CO3的当量由3倍当量增加至5倍当量,碱性增强,有利于催化剂四(三苯基膦)钯(Pd(PPh3)4)的催化活性提高,从而使反应收率得到了提高。化合物13g的合成中,随着反应温度的提高,反应体系中分子之间的碰撞愈加剧烈,有利于反应收率的提高,但当温度升高至95 ℃,化合物12产生了异构现象,并发生了自身的分子内成环反应。化合物11a~11d的合成中,一价铜离子是催化反应发生所必需的。L-抗坏血酸钠作为配体,先将五合水硫酸铜中的二价铜离子还原为一价铜离子,再催化反应的发生。故直接使用碘化亚铜有利于反应收率的提高。
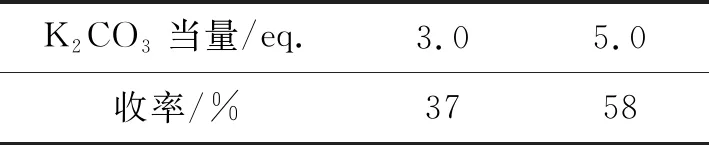
表1 化合物18 优化条件及实验结果

图1 (a)化合物11e 与A2aR的结合模式;(c)分子对接结果二维作用模式;(b)化合物11e 与A2bR的结合模式;(d)分子对接结果二维作用模式

表2 化合物13g 优化条件及实验结果

表3 化合物11a~11d 优化条件及实验结果
2.2 活性测试
采用cAMP法在3种不同浓度下测定了合成的6个化合物对A2a受体的抑制活性并计算抑制率,以AB928做阳性对照,结果见表4。 6个化合物对A2aR受体均表现出不同程度的抑制活性。在1000 nM浓度时,11b和11e的抑制率最强,分别为96.31%、 96.08%。在100 nM浓度时,11b和11f对A2aR受体的抑制率高于阳性药AB928。在10 nM浓度时,11b、11e和11f和均显示出了较好的抑制活性,抑制率均高于阳性药AB928。同时,通过对比抑制率,如11c和11d,初步推断目标化合物中含有吡啶酮结构有利于活性的提高。选取了11b、11e和11f三个目标化合物,分别测定了抑制A2aR和A2bR的IC50值,结果如表5所示。其中,化合物11e抑制A2aR的IC50值(8.188 nM)与阳性药AB928相当,抑制A2bR的IC50值(15.22 nM)明显小于阳性药AB928。

表4 化合物对A2a受体的抑制活性

表5 化合物抑制A2aR和A2bR受体的IC50值
2.3 分子对接
为了明确目标化合物与A2a(PDB ID: 5IU4)、 A2b之间的相互作用,使用Discovery Studio 2017 R2软件对化合物11e进行对接研究。对接结果显示,化合物11e与A2a(40.2565)、A2b(36.2381)对接的CDOCKER INTERACTION ENERGY均高于阳性药物AB928(35.3138),说明化合物11e与A2a和A2b靶点具有较好的亲和作用。化合物11e结构中的氰基、亚甲基除了通过范德华力分别与A2aR的LYS153、GLU169相互作用外,还与LEU249、 VAL84、 ILE274等残基相互作用。另外,化合物11e结构中的氰基、羟基还可以分别与A2bR的ASN254、 THR89通过氢键相互作用提高与靶标的结合能力。
设计并合成了一系列具有吡啶酮或吡唑结构的A2腺苷受体拮抗剂,采用cAMP法研究化合物对A2aR和A2bR受体的抑制活性,并利用分子对接模拟化合物11e对A2a和A2b靶点的结合情况。结果表明:化合物11e对A2aR和A2bR的抑制活性最强,其中对A2bR的抑制性优于阳性对照药AB928。化合物11e是一个具有广泛前景的双靶点(A2a和A2b)腺苷受体拮抗剂,也为小分子免疫检查点抑制剂的研究与开发提供了有价值的借鉴意义。
