甘薯块根响应长喙壳菌(Ceratocystis fimbriata)侵染的转录组测序分析
2022-10-09杨冬静马居奎陈晶伟高方园张成玲孙厚俊谢逸萍
杨冬静,马居奎,唐 伟,陈晶伟,高方园,张成玲,孙厚俊,谢逸萍
(江苏徐淮地区徐州农业科学研究所/农业农村部 甘薯生物学与遗传育种重点实验室,江苏 徐州 221131)
由甘薯长喙壳菌(Ceratocystis fimbriata Ellis et Halsted)引起的甘薯黑斑病(black rot)是甘薯的主要病害之一,在甘薯栽培和贮藏期均可发生;甘薯长喙壳菌分布广泛,种群分化复杂,可侵染甘薯幼苗茎基部和块根,导致死苗、烂床以及烂窖[1]。我国每年由甘薯黑斑病造成的产量损失率达5%~10%,发病严重时可达20%~50%[2]。目前在生产上可采用温汤浸种来防治甘薯黑斑病,但是该方式操作复杂,不适宜推广应用。本课题组历年对全国各地的甘薯品种品系进行黑斑病抗性鉴定工作,迄今尚未发现对甘薯黑斑病免疫且兼具高产、高干率特征的甘薯品种,这类甘薯品种的缺乏也成为疫区甘薯高产优质的主要限制性因素之一。在化学药剂防治方面,张德胜等[3]研究发现粉唑醇、百菌清等药剂对贮藏期薯块黑斑病的防治效果较好,但是粉唑醇残留量高于欧盟规定的最大残留限量标准。
植物病原菌侵染寄主是一个复杂的动态过程,涉及该过程的关键基因及其差异表达引起的重要代谢途径变化从而触发植物的免疫反应一直是人们关注的焦点[4-8]。采用转录组测序分析方法研究植物与病原菌的互作机理,并挖掘重要的抗病基因已有较多的报道[9-13]。笔者以甘薯块根为研究对象,基于转录组测序技术,研究了黑斑病菌侵染过程中甘薯块根在转录组水平上的变化,深入分析了与甘薯响应黑斑病菌侵染过程密切相关的差异基因和代谢途径,并且初步探究了甘薯与黑斑病菌互作过程的抗病分子调控网络,以期挖掘出甘薯与黑斑病菌互作过程中的关键抗病调控因子,为后续研究提供理论依据。
1 材料与方法
1.1 供试材料
供试甘薯品种为相对抗黑斑病品种南京92(R)和相对感病品种徐薯18(S),选取这2个品种的健康、大小均匀的薯块,每个品种取3块,洗净晾干后,采用75%的酒精喷雾杀菌,自然晾干后备用。用灭菌水洗涤在PDA平板上培养5~7 d的甘薯黑斑病菌,制备成分生孢子悬浮液,再采用灭菌纱布过滤掉洗下的菌丝,在显微镜下调节分生孢子悬浮液的浓度为105CFU/mL,备用。
1.2 接种处理方法
用灭菌的针头蘸取分生孢子悬浮液,再针刺薯块,每个薯块均匀接种3排,每排接种5个点;分别于接种0、1、3、7 d后取少量发病部位的薯块,将抗、感品种的各薯块样品分别命名为R1、R2、R3、R4,S1、S2、S3、S4;将各样品用液氮速冻,然后置于-70 ℃冰箱中保存,待各样品收集完成后用干冰寄送至广州基迪奥生物科技有限公司进行测序。
1.3 RNA的提取、检测和文库制备
采用TIANGEN多糖多酚植物总RNA提取试剂盒[天根生化科技(北京)有限公司,货号DP441]提取样品的RNA,具体操作步骤参考该试剂盒的说明书。为了保证测序的质量,通过以下严格的质控检测控制RNA的提取质量:(1)琼脂糖凝胶电泳检测,分析样品RNA的完整性、是否降解,以及其是否存在DNA污染;(2) NanoPhotometer spectrophotometer检测,检测RNA的纯度(OD260/OD280及OD260/OD230比值);(3)Agilent 2100 bioanalyzer检测,利用测定得到的RNA完整值[RNA integrity number (RIN),即RNA分子完整数]来确定RNA的质量,RIN的取值范围为0~10,其值越大表示RNA 的质量越好,越完整。
文库制备方法:用带有Oligo(dT)的磁珠富集具有polyA尾巴的真核mRNA后,再用超声波把mRNA打断。以片段化的mRNA为模板,以随机寡核苷酸为引物,在M-MuLV逆转录酶体系中合成cDNA第一条链;随后用RNaseH降解RNA链,并在DNA polymerase I体系下,以dNTPs为原料合成cDNA第二条链。纯化后的双链cDNA经过末端修复、加A尾并连接测序接头,用AMPure XP beads筛选200 bp左右的cDNA,进行PCR扩增,并再次使用AMPure XP beads纯化PCR产物,最终获得文库。
1.4 数据质控
为了保证数据质量,在信息分析前对原始数据进行过滤,从而减少无效数据带来的分析干扰。首先,对下机的raw reads利用fastp[14]进行质控,过滤掉低质量数据,得到clean reads,reads过滤的步骤如下:去除含adapter的reads;去除含N比例大于10%的reads;去除全部都是A碱基的reads;去除低质量reads(质量值Q≤20的碱基数占整条read的50%以上)。原始数据经过过滤后,分析其碱基的组成及质量分布,以直观展示数据的质量情况。碱基组成越平衡,则质量越高,后续分析越准确。
1.5 序列比对分析
1.5.1 核糖体比对 在正常生物体内,mRNA只占总RNA的很少部分,其他绝大多数都是核糖体RNA(rRNA)。用带有Oligo(dT)的磁珠富集带有polyA尾的mRNA过程中,由于受样品质量和物种的影响,仍可能会有部分rRNA残留。笔者使用短reads比对工具bowtie2[15]将clean reads比对到该物种的核糖体数据库,在不允许错配情况下去除比对上核糖体的reads,将保留下来的unmapped reads用于后续转录组分析。
1.5.2 参考比对 本研究的比对选用甘薯品种泰中6号的参考基因组(http://public-genomes-ngs.molgen.mpg.de/SweetPotato/DOWNLOADS/)。由 于选择比对到基因组,因此所选用的比对软件要求能够进行splice,否则会丢失很多有效的junction reads,这是因为可变剪接和polyA尾在真核生物中普遍存在,跨越可变剪接位点或ployA尾边缘的reads普遍存在。HISAT2软件[16]采用全局和局部搜索的方法能够有效地比对到RNASeq测序数据中的spliced reads,是目前比对率最高且最准确的比对软件。因此本研究采用HISAT2软件开展基于参考基因组的比对分析。
1.6 表达量统计
基因表达量的准确性非常依赖转录本重构结果的完善程度。Stringtie软件[17]能够有效利用转录本的丰度信息,组装出更多的转录本,组装结果也更为准确。根据HISAT2的比对结果,利用Stringtie重构转录本,并计算每个样本中所有基因的表达量。表达量以原始reads count和FPKM展示,其中原始reads count表示该转录本所包含的reads数目,但受测序量和基因长度的影响,原始reads count不利于样本间的差异基因比较。为了保证后续分析的准确性,本文先对测序深度进行校正,再对基因或转录本的长度进行校正,在获得基因的FPKM值后,再进行后续分析。
1.7 样本关系的主成分分析
基于表达量信息,利用R(http://www.r-proje-ct.org/)开展主成分分析(Principal Component Analysis,PCA),从而利用降维的思路研究样本间的距离关系。在所得的分析结果中,样品组成越相似,则在PCA图中的距离越近;来自不同有效处理的样品往往表现出各自聚集的分布。
1.8 差异表达基因(DEGs)分析
基因差异表达分析的输入数据为基因表达水平分析中得到的reads count数据,使用DESeq2软件[18]进行差异表达基因的分析,主要分为3个步骤:对reads count数据进行标准化(normalization);根据模型进行假设检验概率(P value)的计算;最后进行多重假设检验校正,得到FDR值(错误发现率)。本研究以FDR<0.05且|log2FC|>1为标准筛选差异表达基因,然后进行统计分析。
1.8.1 GO富 集 分 析 Gene Ontology(简 称GO)是一个国际标准化的基因功能分类体系,其提供了一套动态更新的标准词汇表(controlled vocabulary)来全面描述生物体中基因和基因产物的属性。GO总共有3个ontology(本体),分别描述基因的分子功能(molecular function)、细胞组分(cellular comp-onent)、参与的生物过程(biological process)。GO的基本单位是term(词条、节点),每个term都对应1个属性。GO功能分析一方面给出基因的GO功能分类注释,另一方面给出基因的GO功能显著性富集分析。在进行GO富集分析时,首先将基因向GO数据库(http://www.geneontology.org/)的各个term映射,并计算每个term的基因数,从而得到具有某个GO功能的基因列表及基因数目;然后应用超几何检验,找出与整个基因组背景相比在基因中显著富集的GO条目。
1.8.2 KEGG富集分析 在生物体内,不同的基因相互协调,行使其生物学功能,基于Pathway的分析有助于进一步了解基因的生物学功能。KEGG是有关Pathway的主要公共数据库。Pathway显著性富集分析以KEGG Pathway为单位,应用超几何检验,找出与整个基因组背景相比在基因中显著富集的Pathway。通过Pathway显著富集分析能确定基因参与的最主要生化代谢途径和信号转导途径[19]。
1.9 植物抗病相关基因(R-gene)分析
将预测的蛋白序列与相应的R-Gene数据库(PRGdb v3.0,http://prgdb.org)进行hmmscan 比对,筛选与植物抗病相关的基因[20]。
2 结果与分析
2.1 抗、感甘薯品种接种黑斑病菌的表型差异
抗、感甘薯品种在接种甘薯黑斑病菌分生孢子悬浮液后,分别于第1、3、7天取出薯块,进行表皮和剖面观察,并拍照记录发病表型差异。从图1可以看出:薯块被侵染的第1天,抗、感品种的表皮和剖面均未出现明显的发病症状;薯块被侵染的第3天,抗、感品种的表皮和剖面均表现出明显的黑斑症状,但在表型上并无显著差异;薯块被侵染的第7天,抗、感品种的表皮均表现出明显的黑斑症状,病斑直径差异不显著,但从剖面来看,感病品种被侵染的深度明显比抗病品种深,且感病品种与抗病品种的发病体积呈显著差异。
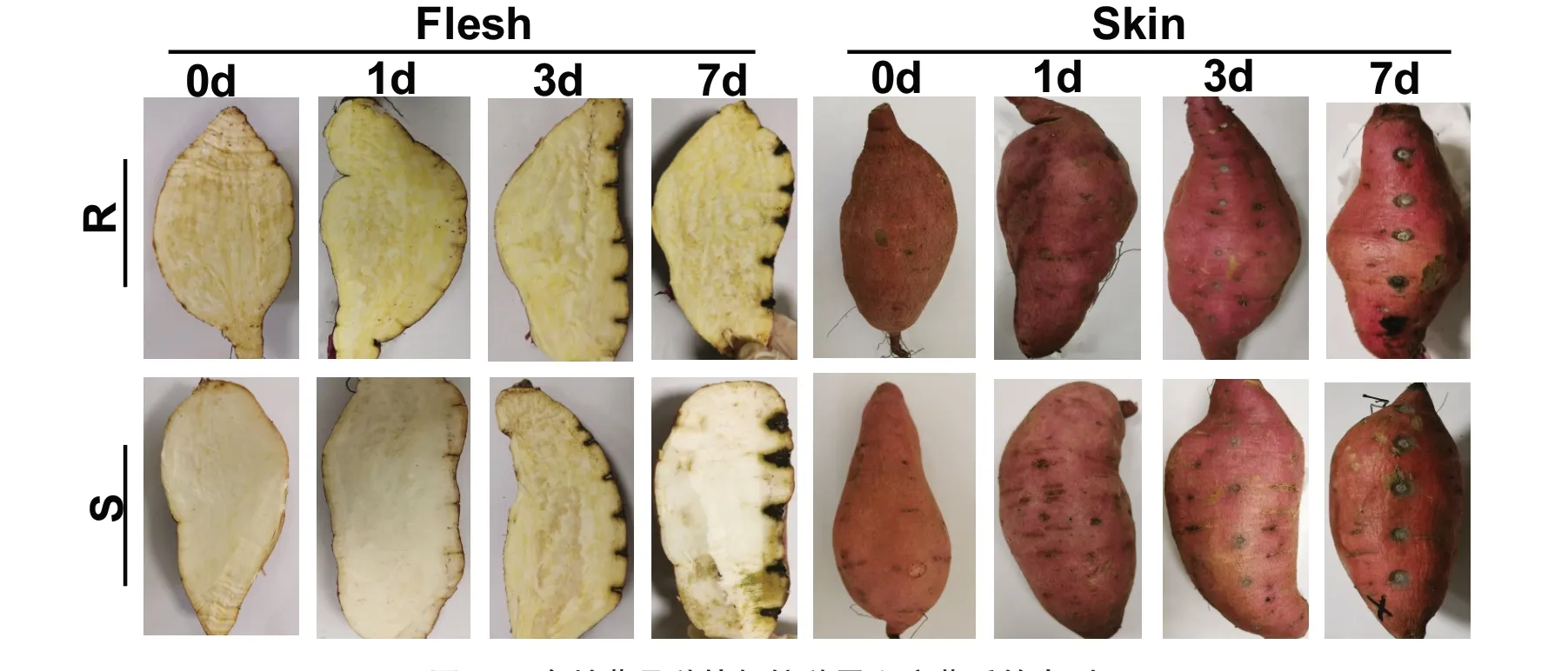
图1 2个甘薯品种块根接种黑斑病菌后的表型
2.2 总RNA提取和质量分析
基于抗、感品种接种黑斑病菌后的表型差异,分别在不同侵染时间点取样并提取RNA,琼脂糖凝胶电泳检测结果显示,本研究中24个样品的RNA完整性较好,未发现明显降解,且不存在DNA污染。此外,采用RNA纯度和浓度检测的结果也显示所提取的RNA质量完全符合建库和转录组测序的要求。
2.3 测序质控
对原始数据进行质量控制以后,获得的clean reads数目统计结果如表1所示,各样品的clean reads数目均在62529524~88661116。碱基测序错误率分析显示:Q20碱基百分比(错误率<1.0%)分布介于97.51%~98.33%,Q30碱基百分比(错误率<0.1%)分布介于92.87%~95.08%,GC含量介于56.40%~77.77%。以上结果均表明测序数据的质量较高,过滤后的有效序列reads可用于后续生物信息学分析。
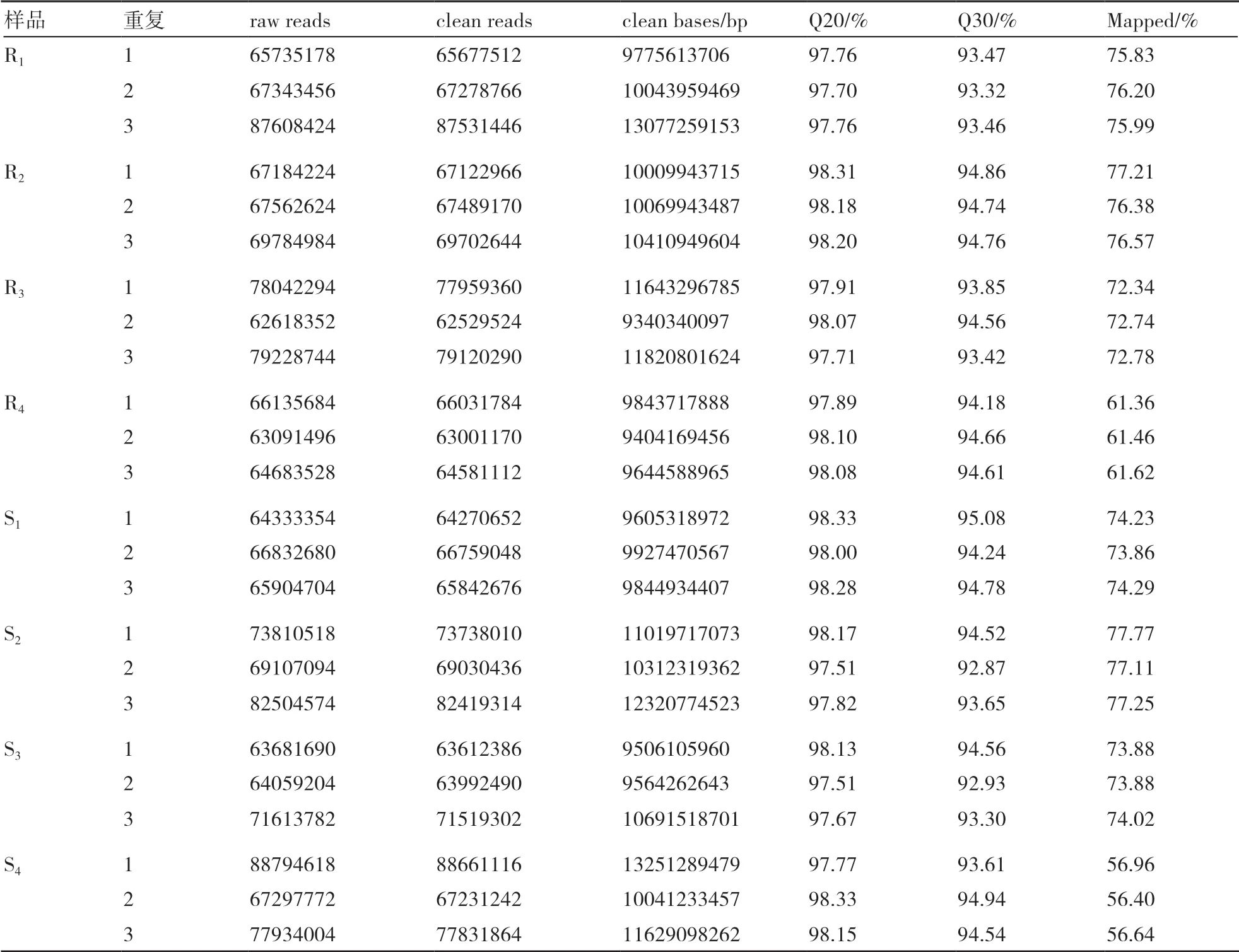
表1 转录组测序数据统计分析结果
2.4 主成分分析
各样本基因表达值(FPKM)的主成分分析结果如图2所示,在相同时间点组内样品的表达基因在每个生物学重复中都比较接近(S4除外),抗病品种的第0天对照组R1与第1、3和7天的处理组R2、R3、R4明显比较远,且处理时间越长,各处理组与R1的距离越远。抗病处理组R2虽然在症状上表现不明显,但是随着黑斑病菌的侵染其差异表达基因的类别和表达量都有了较大的变化,故而与对照组R1差异较大。随着侵染时间的增加,差异表达基因的类别和表达量变化更多,但与R1相比R4、R3已经很接近,而侵染7 d时的症状表型差异更明显,这表明差异基因的表达和系列防卫反应发生在侵染早期。感病品种4个组(S1~S4)的基因表达规律与抗病品种相似,不同的是感病品种的对照组S1与处理组S2、S3、S4之间的距离更近,表明随着病原菌侵染时间的延长,感病品种的表达基因类别和表达量也有变化,但是变化幅度小于抗病品种。从图3还可以看出,感病品种S1与抗病品种R1之间的距离较大,表明在两者之间基因类别和表达量差异也非常明显。
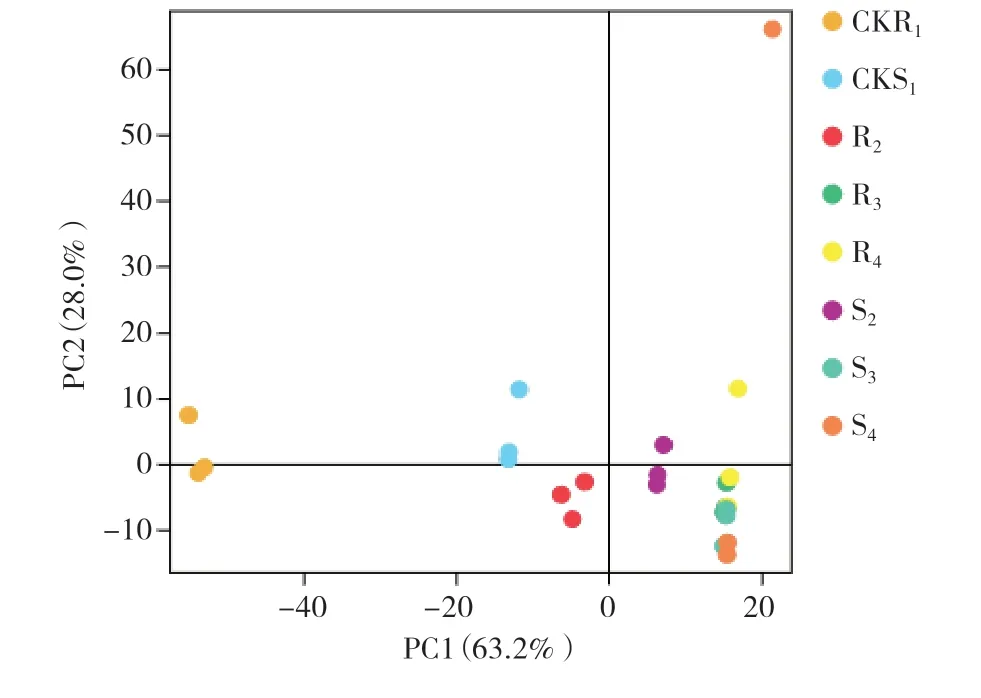
图2 各样本的主成分分析结果
2.5 抗、感品种中差异表达基因的统计分析
基于差异分析的结果,筛选FDR<0.05并且|log2FC|>1的基因为显著差异基因。由图3可见,与抗病品种对照组R1相比,处理组R2有4771个差异基因上调表达,2423个基因下调表达;处理组R3有6405个差异基因上调表达,7725个基因下调表达;处理组R4的上调表达差异基因数比R3略少,下调表达的差异基因数比R3增多;总的来看,随着侵染时间的增加,差异基因数呈上升趋势。感病品种中差异基因上调和下调表达的变化规律与抗病品种相似,也是随着侵染时间的增加,差异表达的基因数增多,且均在侵染第3天的增幅最大。比较同一取样时间点抗病品种和感病品种的基因表达情况,发现随着侵染时间的增加,抗、感品种的差异表达基因数逐渐减少,差异表达基因数从0 d的12124个逐渐下降到7 d的3468个。

图3 不同处理组间差异表达基因统计结果
抗病品种3个处理组与对照组对比的维恩图(图4A)表明,在第1、3、7天均出现差异表达的基因有4955个。感病品种3个处理组与对照组对比的维恩图(图4B)显示,在第1、3、7天均出现差异表达的基因有5644个。从图4C可以看出,参与响应黑斑病菌侵染的核心差异基因有2524个。从相同侵染时间点来分析抗、感品种之间的差异,发现在R1vs S1中,R1上调表达的基因数为5290,下调表达的基因数为6834,表明在侵染之前抗、感品种之间就存在了差异,且随着侵染时间的增加,抗、感品种之间上调和下调表达的基因数均逐渐减少;在R1vs S1、R2vs S2、R3vs S3和R4vs S4这4个对比组中有1057个共有的差异表达基因(图4D),这些共有的DEGs与侵染时间点没有关系,是抗、感品种之间本身就存在的差异,与黑斑病菌侵染与否无关。
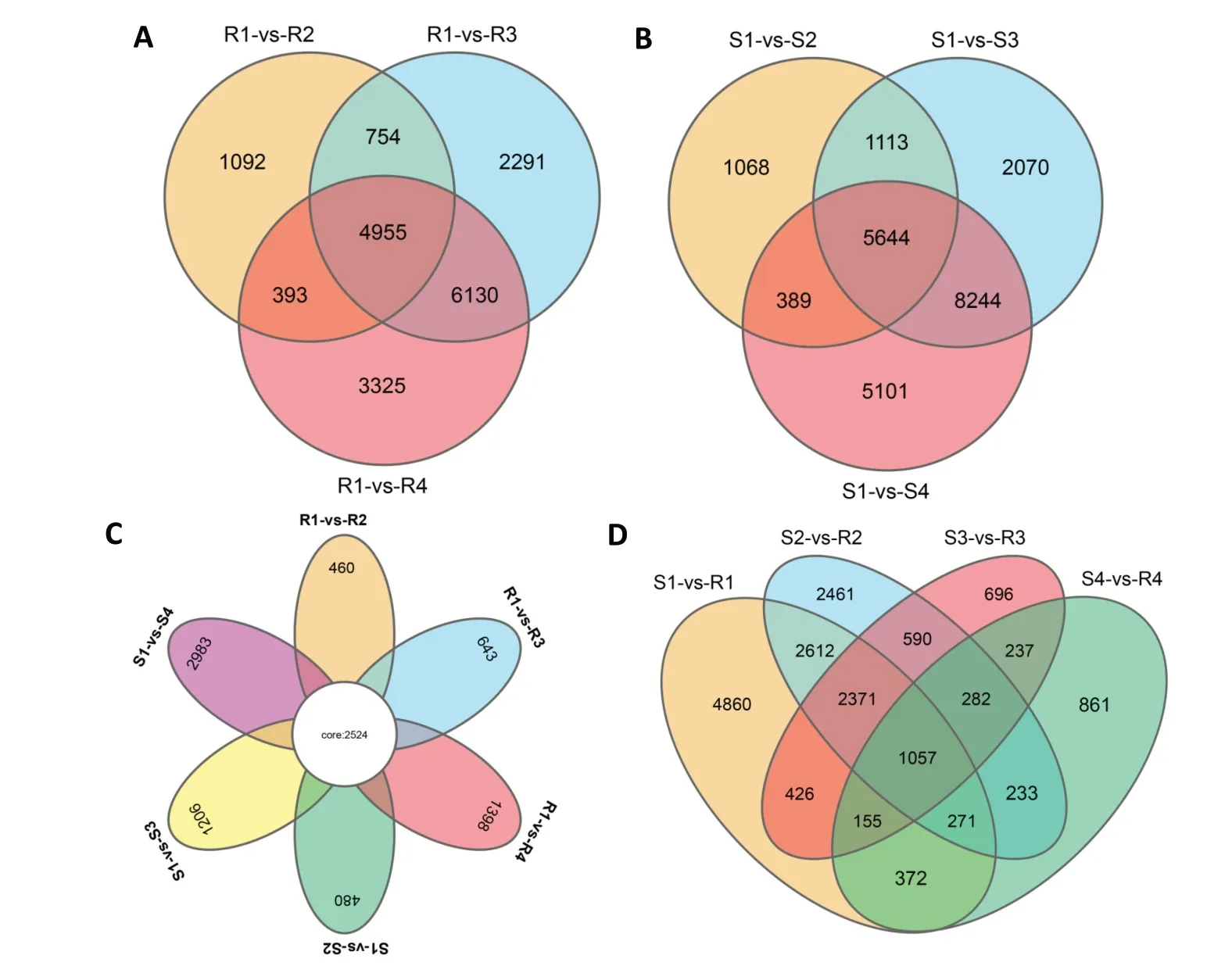
图4 抗、感甘薯品种差异表达基因的维恩分析结果
2.6 差异基因GO功能的显著性富集分析
选取侵染3 d的处理组与对照组相比,进行差异基因GO(Gene Ontology)功能的显著性富集分析,筛选了显著富集前20的GO条目,GO富集圈结果如图5所示。抗病品种被侵染3 d的差异基因中,可明显观察到隶属于生物过程的是单一生物代谢过程(GO:0044710)、前体代谢产物和能量的产生(GO:0006091)和对金属离子的响应(GO:0010038);分子功能的GO富集分析显示,主要是氧化还原酶活性(GO:0016491)和四吡咯结合(GO:0046906);细胞组分的GO富集分析显示,主要包括细胞膜(GO:0016020)、膜的固有成分(GO:0031224)和质体(GO:0009536)等15个条目(图5A)。对感病品种侵染3 d的差异基因进行GO富集分析,结果(图5B)显示,在被富集到的前20的GO条目中,有4个条目(GO:0044710等)是参与生物过程的;有3个条目(GO:0016491等)是参与分子功能的;有13个条目(GO:0009536等)均是参与细胞组分相关功能的。对比图5A和图5B可以看出,隶属于生物过程的单一生物代谢过程(GO:0044710)、隶属于分子功能的氧化还原酶活性(GO:0016491)和四吡咯结合(GO:0046906),以及隶属于细胞组分的质体(GO:0009536)和细胞外围(GO:00071944)等条目在抗、感品种中均被富集到,表明当长喙壳菌侵染甘薯块根时抗、感品种在这些生物过程中均有响应。
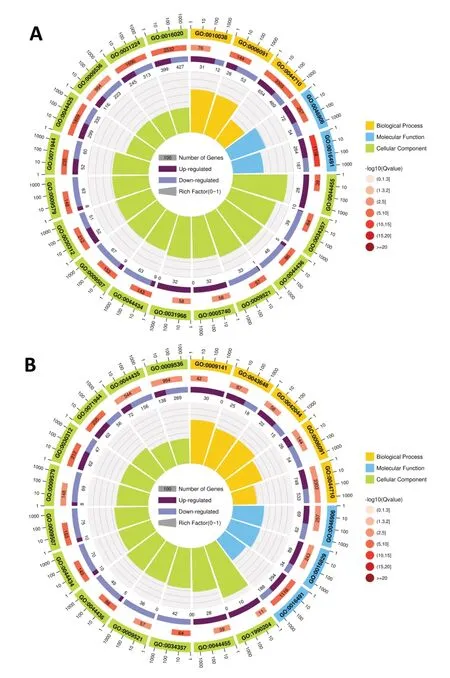
图5 抗病及感病品种中对照组和处理组的差异GO富集圈分析结果
此外,还有一些GO条目是在抗病品种中富集到而在感病品种中没有富集到的,如隶属于细胞组分的膜的固有成分(GO:0031224)和细胞膜部件(GO:0044425),这些过程可能是抗病品种与感病品种抵御长喙壳菌侵染的差异所在。
2.7 差异基因的KEGG通路富集分析
图6A和图6B分别为在抗病甘薯块根R1vs R3对比组和感病甘薯块根S1vs S3对比组中,差异基因的功能主要富集到的前20的KEGG通路。从中可以看出,抗、感品种中的差异基因均被富集到的KEGG通路包括代谢途径、次级代谢产物的生物合成、碳代谢、苯丙烷生物合成、柠檬酸循环/三羧酸循环、光合作用、氧化磷酸化、谷胱甘肽代谢、氨基酸生物合成、糖酵解/糖异生、光合作用天线蛋白、光合生物的固碳作用、托烷、哌啶和吡啶生物碱的生物合成以及果糖和甘露糖代谢等14个通路;与感病品种不同,抗病品种中的差异基因还被富集到植物激素信号转导、淀粉和糖代谢、萜类主链生物合成、氨基糖和核苷酸糖代谢、戊糖磷酸途径,以及ABC转运蛋白等信号通路;与抗病品种不同,感病品种中的差异基因还被富集到植物MAPK信号通路、半胱氨酸和蛋氨酸代谢、乙醛酸和二羧酸代谢、甘氨酸、丝氨酸和苏氨酸代谢、二苯乙烯类、二芳基庚烷类和姜辣素生物合成以及苯丙氨酸代谢途径。
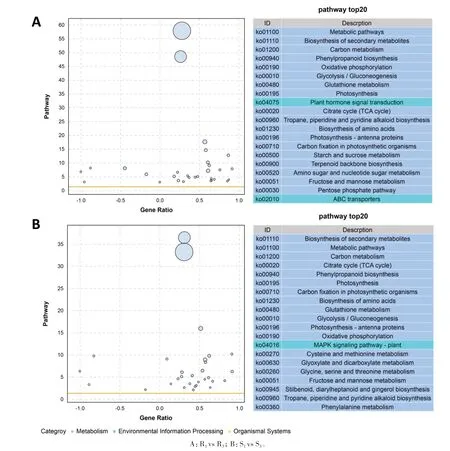
图6 差异表达基因显著富集的前20的KEGG通路
进一步对抗、感品种间4个对比组中1057个共有的差异表达基因进行KEGG通路富集分析,结果如表2所示,从中可以看出,被富集到的前20的KEGG通路包括单萜生物合成、亚油酸代谢、氨基糖和核苷酸糖代谢、剪接体、异喹啉生物碱生物合成、核苷酸切除修复、其他聚糖降解、酪氨酸代谢、错配修复、泛素介导的蛋白质水解、自噬-其他真核生物、硒化合物代谢、植物激素信号转导、DNA复制、卟啉与叶绿素代谢、RNA降解、核糖体代谢、果糖和甘露糖代谢、氨基酸生物合成以及同源重组等通路,共涉及158个关键抗性相关基因。
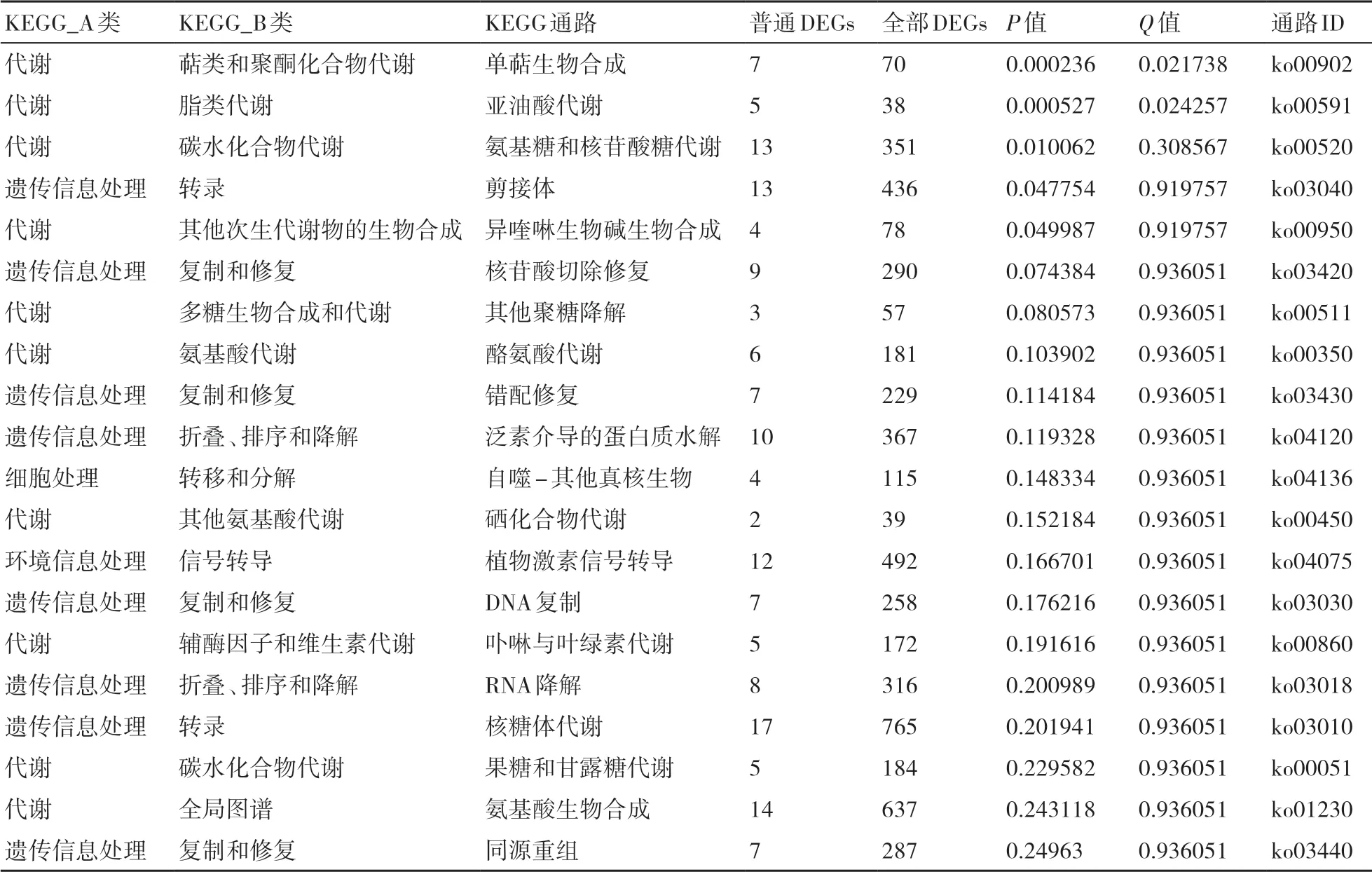
表2 基于各时间点抗、感品种共同响应长喙壳菌侵染的差异基因的前20的KEGG通路
3 讨论和结论
近年来,植物-病原菌互作研究逐渐成为热点,以多种植物为寄主的长喙壳菌以及甘薯栽培品种泰中6号的全基因组序列均已公布,以全基因组序列为依据开展植物抗病机制和病原菌致病机制的研究越来越多,但迄今在国内外尚未见有关长喙壳菌侵染甘薯的转录组测序和分析的研究报道。笔者选择对黑斑病抗性表现不同的2个甘薯品种,研究了长喙壳菌侵染的转录组测序,分析了抗、感品种间本身存在的差异基因及其参与抗病相关途径的差异,所得研究结果有助于揭示不同抗性甘薯品种抗性差异的原因。本文还分析了长喙壳菌侵染过程中抗病和感病品种响应病原菌的差异,揭示了抗、感品种抵御病原菌侵染过程中存在的差异变化。在本研究设置的侵染时间1、3和7 d下,接种的甘薯薯块分别未见明显症状、已初步显示病症、黑斑病症状非常明显,这3个时间点分别是长喙壳菌侵染甘薯的早期、中期和后期。因此,本研究在上述3个时间点取样进行转录组测序,测序结果能全面反映长喙壳菌侵染状态下抗、感甘薯品种的转录本差异,可以为黑斑病抗病调控网络和抗性基因的挖掘奠定基础。在本研究中,RNA-seq数据质控的碱基质量、测序饱和度、组内样品重复性等质量评估结果都比较理想。在与参考基因组的比对中,各处理的clean reads数均在9340340097 bp以上,因此基于该转录组测序数据的后续分析和实验结果均具有较好的真实性和可靠性。
植物响应病原菌侵染及其对病原菌的防御机制是复杂的新陈代谢过程,对病原物侵入及扩展的生理反应是以酶的催化活动来实现的[22]。超氧化物歧化酶、过氧化物酶及过氧化氢酶等是植物体内重要的保护酶类,在保护植物细胞膜免受自由基伤害的过程中起非常重要的作用[23]。本研究通过对南京92和徐薯18转录组测序分析发现,在甘薯长喙壳菌侵染条件下,2个材料中均有大量差异基因被富集到属于分子功能的与氧化还原酶活性相关的GO条目(GO:0016491)中,南京92和徐薯18中分别有451和482个DEGs,表明在长喙壳菌侵染下抗、感品种体内的保护酶系统平衡状态均被打破,从而抵御长喙壳菌的侵染,维持机体的正常运转;同时,长喙壳菌侵染甘薯是复杂的生物过程,在此过程中差异基因被富集到属于生物过程的单一生物代谢GO条目(GO:0044710),抗、感品种中分别有1123和1281个DEGs,从富集的DEGs数量来看,南京92受长喙壳菌侵染的影响比徐薯18小。
KEGG富集分析结果显示,抗、感品种中的差异基因均被富集到的前20的KEGG通路包括代谢途径、次级代谢产物的生物合成、碳代谢、苯丙烷生物合成、柠檬酸循环/三羧酸循环、光合作用、氧化磷酸化、谷胱甘肽代谢等,这表明抗、感品种在抵御长喙壳菌侵染过程中有相似的反应。另外,在抗、感品种间的R1vs S1、R2vs S2、R3vs S3和R4vs S4四个对比组中发现了1057个共有的差异表达基因,这些共有的DEGs与黑斑病菌侵染没有关系。此外,通过R-gene分析,鉴定到221个抗性相关基因,这些抗病基因主要为激酶类基因(图7)。
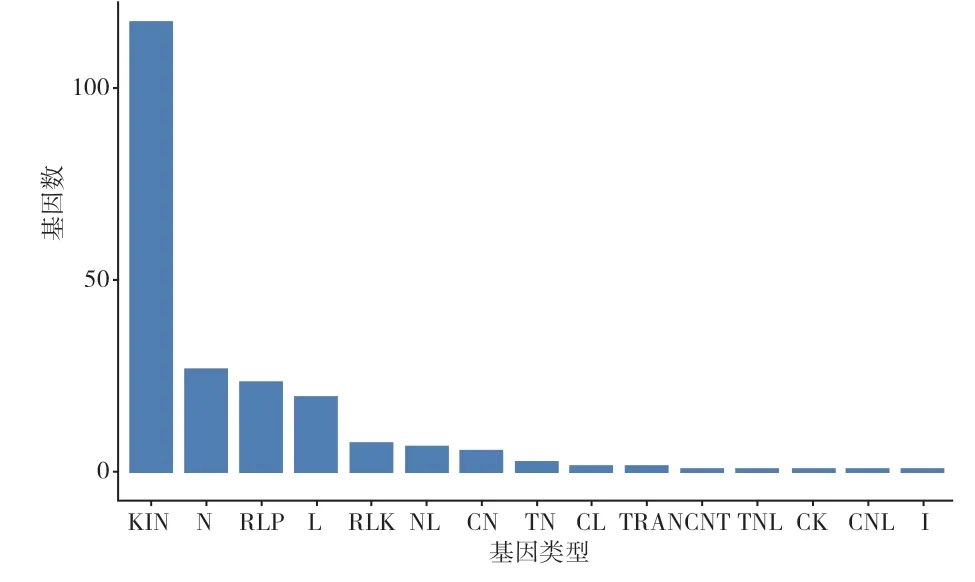
图7 响应长喙壳菌侵染的甘薯R-gene分析结果
本研究通过对甘薯长喙壳菌侵染甘薯块根的转录组测序和分析,发现了甘薯在长喙壳菌侵染条件下的大量DEGs;通过GO和KEGG富集分析,初步明确了在长喙壳菌侵染条件下甘薯的生物学过程变化以及差异基因被富集到的主要通路;此外还鉴定出了一些抗性相关基因。后续拟结合基因注释和富集分析结果,对重点关注的相关基因进行克隆和分析,并对这些基因表达的蛋白质功能和调控网络进行深入研究,以进一步解析甘薯响应长喙壳菌侵染的分子机制。
