RAW264.7细胞MyD88基因缺失株构建①
2022-10-07董明鑫孙成彪张剑旭刘文森
王 燕 陈 为 董明鑫 孙成彪 张剑旭 常 影 刘文森 许 娜③
(中国农业科学院长春兽医研究所,长春 130122)
近年随着“魔剪”CRISPR/Cas9技术崛起,基因编辑在生物医药研究领域掀起了一股研究热潮[1]。与传统基因编辑技术(如锌指核酸酶、活因子样效应物核酸酶等)相比,CRISPR/Cas9技术采用特殊小向导RNA(single guide RNA,sgRNA)靶向目标基因,具有操作简便、靶向效应强等优点,成为当前基因研究领域的有力工具,已广泛用于目的基因筛选编辑和药物靶点筛选等领域[2-3]。髓样分化因子88(myeloid-differentiation primary response protein 88,MyD88)是Toll样受体(Toll-like receptors,TLRs)信号转导中的重要接头分子,其介导的TLRs信号转导称为MyD88依赖型途径,以形成MyD88、IRAK4和IRAK1复合体为标志,早期活化NF-κB和MAPK为特征[4]。近年研究发现,MyD88不仅在固有免疫中发挥重要作用,还可调控适应性免疫,是连接二者的桥梁,但部分机制尚不明确[5]。为深入开展MyD88功能及相关调控机制研究,本研究拟采用CRISPR/Cas9技术定点敲除MyD88基因,制备MyD88缺失的稳定RAW264.7细胞株。
1 材料与方法
1.1 材料293T细胞株、RAW264.7细胞株、大肠杆菌菌株DH5-α均为本实验室保存;三质粒系统(pSPAX2、pMD2G和穿梭质粒)、pHBLV-U6-gRNAEF1-ZsGreen载体(汉恒);质粒DNA抽提试剂盒(Axygen);限制性内切酶、T4连接酶(TaKaRa);LipofiterTM转染试剂盒、胎牛血清、1640培养液、0.25%胰酶、Puromycin(Life);RNA提取试剂盒(奕杉);MyD88抗体(Cell Signaling Technology);羊抗兔IgG抗体(Proteintech);Luminata forte western HRP substrate(Merck Millipore);脂多糖LPS、肽聚糖PGN(Sigma Aldrich);Mouse TNF-αELISA kit(达优);CO2细胞培养箱、低温高速离心机(Thermo);荧光显微镜(Olympus);荧光定量PCR仪(ABI);高端凝胶成像系统(Alpha Innotech);高端多功能酶标仪(TECAN);参照NCBI中目的基因序列,应用Primer 5.0软件设计引物(表1),引物由上海生工生物公司合成;TNF-α引物购自GeneCopoeia。
1.2 方法
1.2.1 HBLV-Cas9-PURO慢病毒包装和滴度测定将pSPAX2、pMD2G和携带Cas9基因的穿梭质粒进行扩增,经大量高纯度无内毒素抽提后,共转染293T细胞,6 h后更换为完全培养液培养48 h和72 h,收集富含慢病毒颗粒的细胞上清,4℃、2 000 g离心10 min,去除细胞碎片,收集病毒上清,离心120 min,得到高滴度慢病毒超离液(HBLV-Cas9-PURO)。分装病毒、标记、-80℃保存。稀释计数法测定病毒滴度(TU/ml)=细胞数×阳性克隆百分比×MOI×病毒稀释倍数×103。
1.2.2 HBLV-Cas9-PURO感染、筛选及鉴定感染前1 d将RAW264.7细胞适当铺板,细胞密度生长至30%~50%时,将HBLV-Cas9-PURO以MOI=20进行1/2小体积感染,4 h后补足至完全培养体积,24 h后更换新鲜完全培养液,48 h后荧光显微镜初步观察GFP表达效率并更换含10µg/ml Puromycin的新鲜完全培养液进行Puromycin抗性筛选,进行稳定株筛选及扩增,提取细胞总RNA,PCR检测Cas9基因表达,琼脂糖凝胶电泳检测扩增结果。PCR反应条件:94℃10 min,94℃30 s,60℃30 s,72℃30 s,30个循环。
1.2.3 m-MyD88 gRNA重组载体构建和慢病毒包装根据小鼠MyD88在NCBI的基因序列,为实现CRISPR/Cas9打靶载体用于小鼠MyD88基因敲除,设计3条sgRNA序列:m-MyD88 gRNA1、m-MyD88 gRNA2、m-MyD88 gRNA3(表2),由长春库美公司合成后,退火成双链Oligo序列,T4连接酶接入含ZsGreen荧光标记的pHBLV-U6-gRNA-EF1-ZsGreen线性化表达载体,连接产物转化至DH5α感受态细胞,Amp抗性平板培养过夜,挑菌并扩大培养,菌液进行测序。将测序验证正确的阳性克隆进行质粒提取和慢病毒包装,慢病毒(HBLV-m-MyD88 gRNA1-GFP、HBLV-m-MyD88 gRNA2-GFP、HBLV-m-MyD88 gRNA3-GFP和HBLV-GFP NC对照)包装及滴度测定方法同1.2.1。
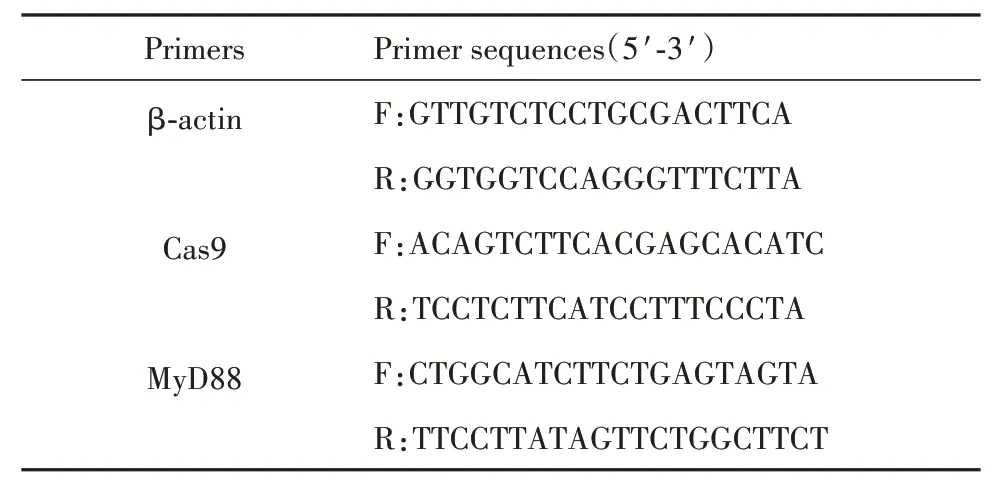
表1 引物序列Tab.1 Primer sequences
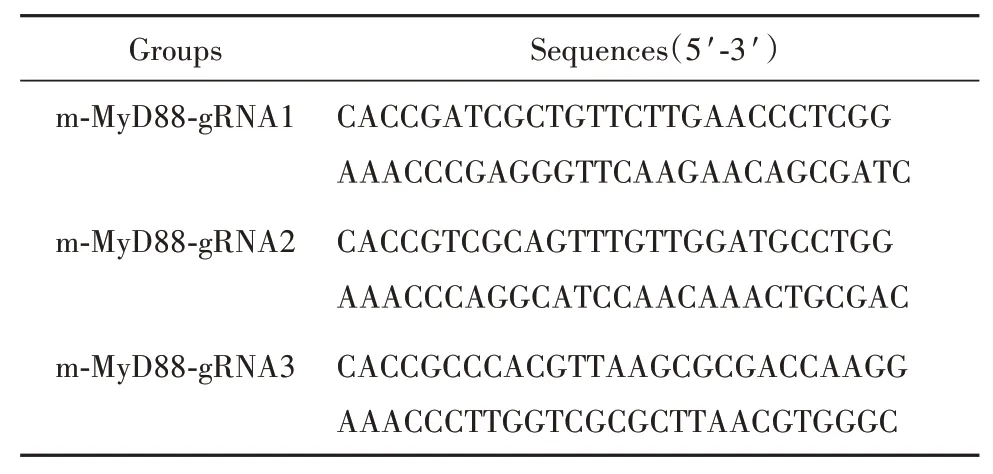
表2 sgRNA寡核苷酸序列Tab.2 Sequences of sgRNA oligonucleotides
1.2.4 HBLV-m-MyD88 gRNA-GFP感染及基因敲除效果鉴定筛选成功的RAW264.7-Cas9细胞以5×105个/ml铺于6孔板,待细胞密度生长至30%~50%融合时,将包装的各组慢病毒以MOI=30进行1/2小体积感染,感染后筛选过程及PCR鉴定方法同1.2.2。同时进行Western blot鉴定,提取感染后的RAW264.7-Cas9细胞蛋白,BCA定量后进行SDS-PAGE电泳,电泳结束后转至PVDF膜,5%牛血清白蛋白(BSA)室温封闭1 h,加入抗MyD88抗体(1∶1 000)4℃孵育过夜,PBST洗膜3次,5 min/次,加入二抗室温孵育1 h,PBST洗膜3次,5 min/次,滴加发光液,高端凝胶成像系统检测MyD88表达。
1.2.5 MyD88基因敲除后TNF-α表达检测为验证敲除MyD88基因的RAW264.7细胞功能,采用MyD88基因激活剂LPS和PGN诱导敲除MyD88基因的RAW264.7细胞8 h,收集上清,ELISA试剂盒检测TNF-α分泌情况;提取细胞总RNA、逆转录、荧光定量PCR检测TNF-α表达。
1.3 统计学处理采用SPSS17.0软件进行数据分析,组间比较采用单因素方差分析,P<0.05为差异有统计学意义。
2 结果
2.1 慢病毒滴度测定采用稀释计数法对构建的慢病毒HBLV-Cas9-PURO、HBLV-GFP NC对照、HBLV-m-MyD88 gRNA1-GFP、HBLV-m-MyD88 gRNA2-GFP、HBLV-m-MyD88 gRNA3-GFP进行滴度测定,测定结果见表3。
2.2 RAW264.7-Cas9细胞株鉴定将HBLV-Cas9-PURO感染并筛选后的细胞进行扩增,提取正常组和感染后的RAW264.7-Cas9细胞RNA,PCR检测Cas9基因表达。与正常RAW264.7细胞比较,感染后Cas9基因扩增产物表达升高(图1),表明获得稳定表达Cas9基因的RAW264.7细胞(RAW264.7-Cas9)。
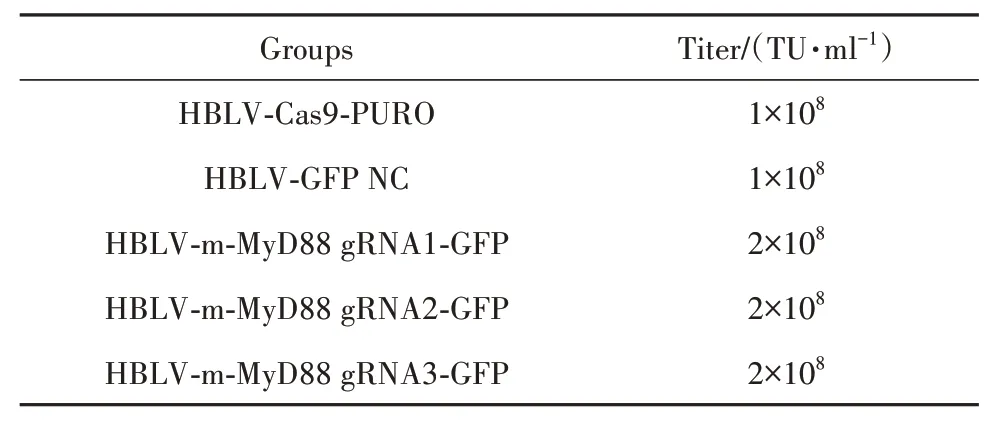
表3 慢病毒滴度测定结果Tab.3 Results of lentivirus titer determination
2.3 gRNA重组载体测序结果 将单链sMyD88-gRNAs退火成双链Oligo序列,连接入单酶切线性化的载体pHBLV-U6-gRNA-EF1-ZsGreen。转化子由上海汉恒生物科技公司进行测序(图2),插入序列位置和方向与预期相符,证明3个载体构建成功。
2.4 RAW264.7-Cas9细胞感染及敲除效果鉴定各组慢病毒分别感染RAW264.7-Cas9后,荧光显微镜下观察gRNA重组载体导入情况,结果显示目的基因导入成功(图3)。PCR检测各组细胞MyD88基因表达,与对照组比较,HBLV-m-MyD88 gRNA3-GFP感染后RAW264.7-Cas9细胞MyD88基因表达显著下调(图4A)。为进一步验证敲除效果,采用Western blot检测MyD88蛋白表达,结果显示,HBLV-m-MyD88 gRNA1-GFP和HBLV-m-MyD88 gRNA3-GFP感染后细胞MyD88蛋白表达均下降,且HBLV-m-MyD88 gRNA3-GFP组下降最为显著(图4B)。说明MyD88基因敲除成功,gRNA3组敲除效果最佳。
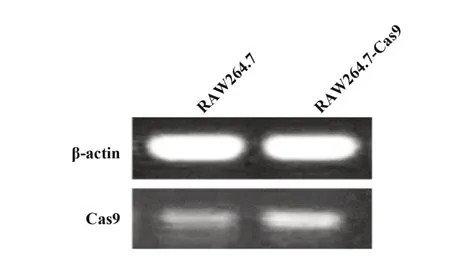
图1 Cas9基因PCR结果Fig.1 PCR analysis of Cas9 gene

图2 MyD88-gRNA测序图Fig.2 Sequencing data of MyD88-gRNA
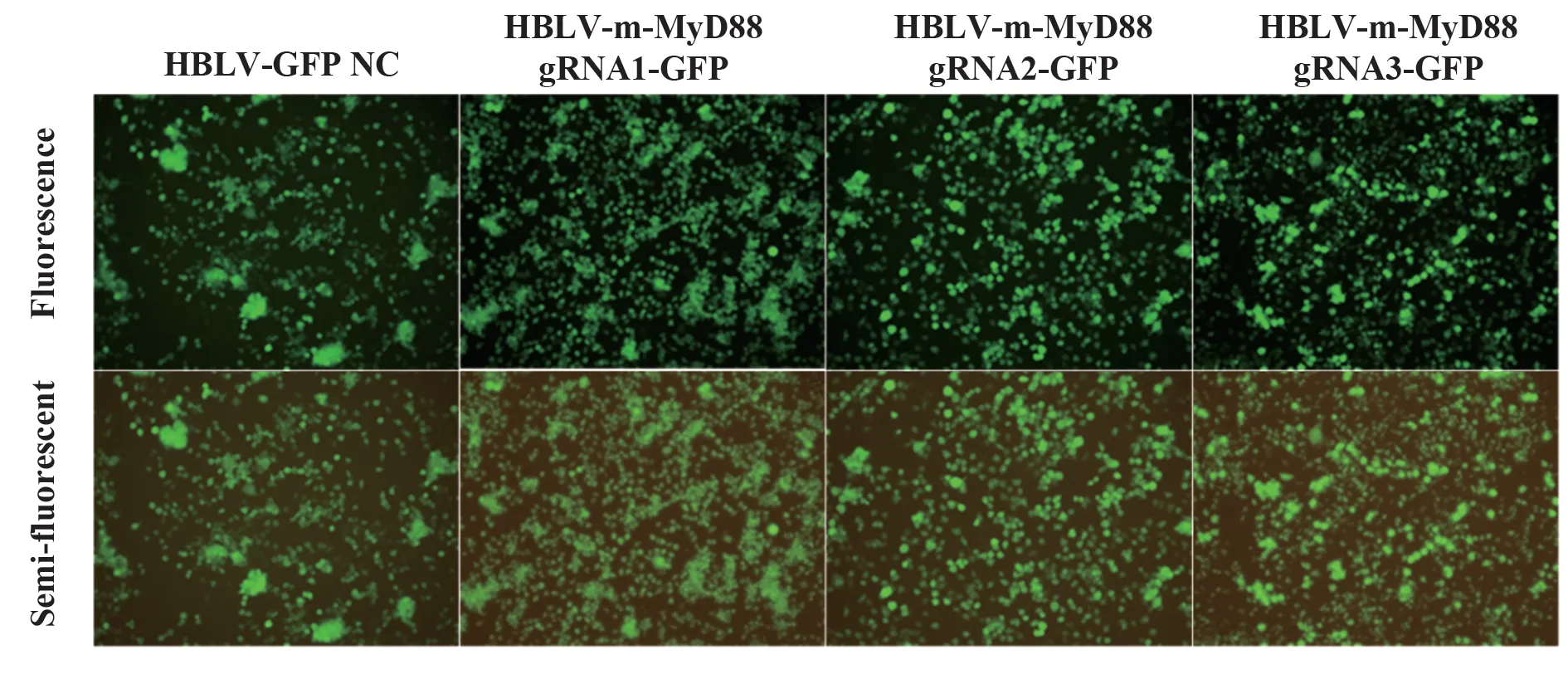
图3 荧光显微镜下视野(×10)Fig.3 Field of vision under fluorescence microscope(×10)
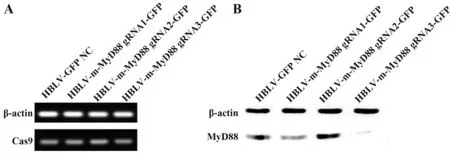
图4 MyD88基因敲除后PCR扩增产物及蛋白表达Fig.4 Expressions of PCR products and protein after MyD88 gene knockout
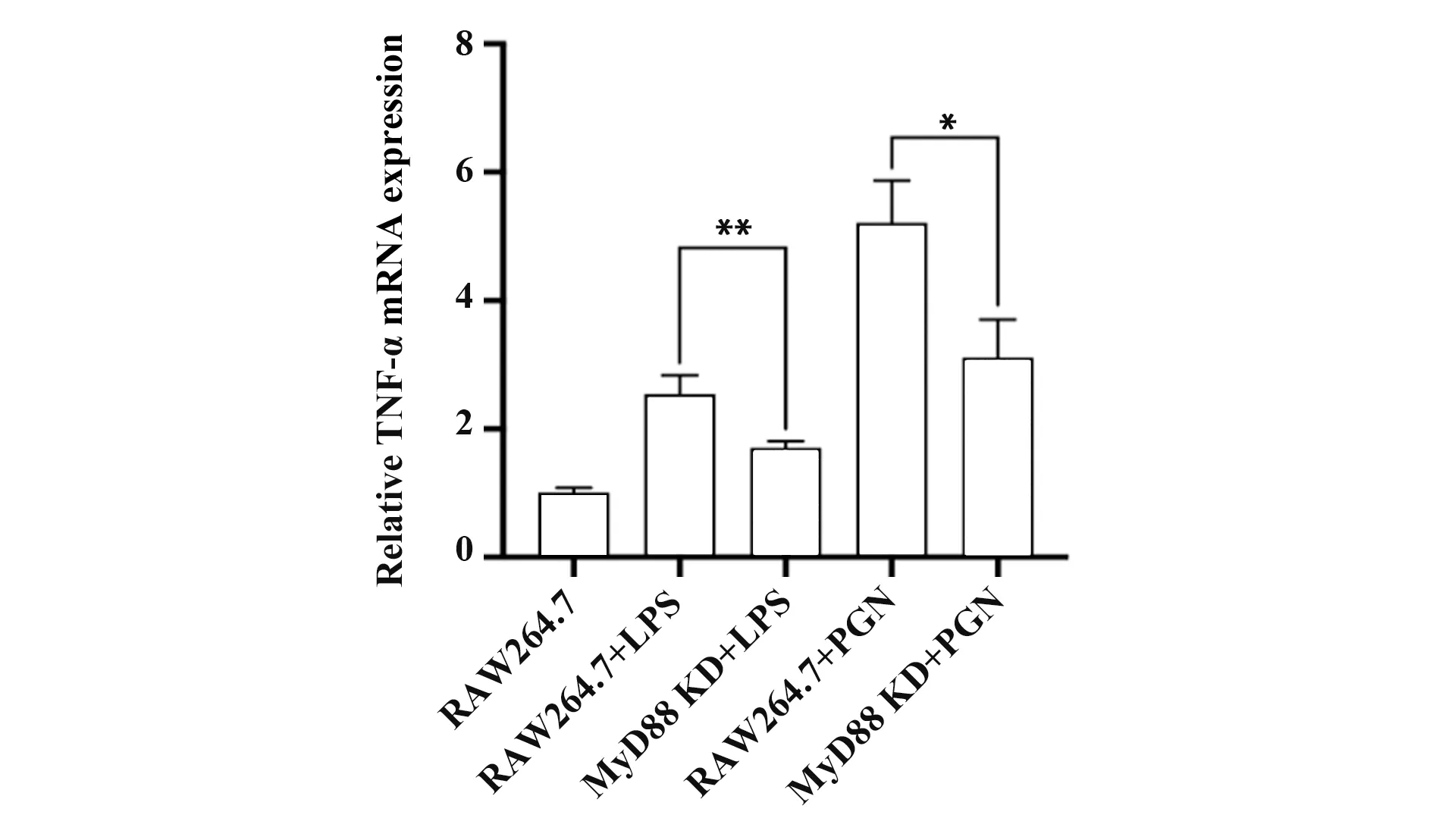
图5 MyD88基因敲除后TNF-αmRNA表达Fig.5 Expression of TNF-αmRNA after MyD88 knock down
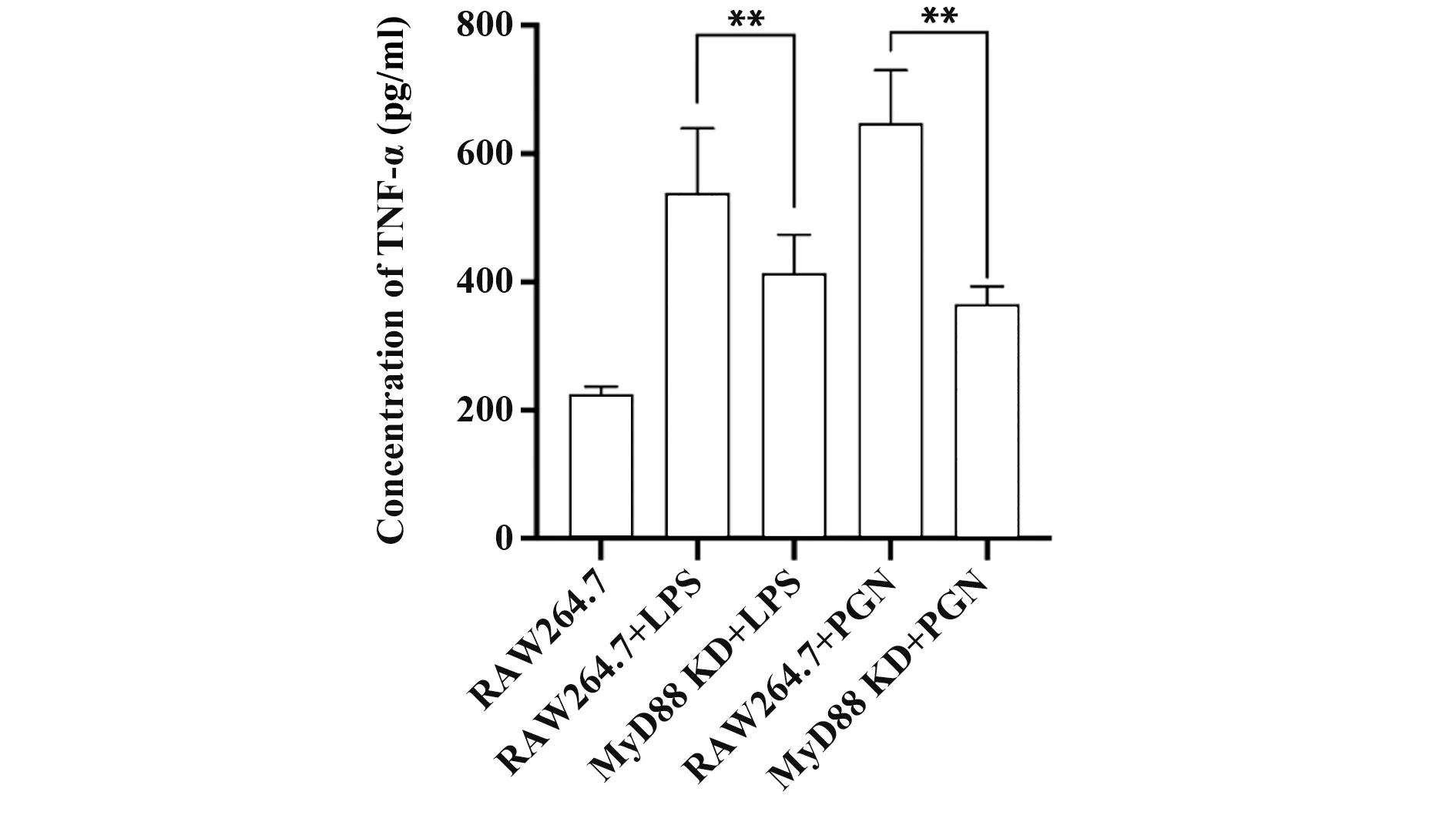
图6 MyD88基因敲除后细胞上清TNF-α分泌水平Fig.6 Secretion level of TNF-αafter MyD88 knock down
2.5 MyD88基因敲除后TNF-α水平检测 采用MyD88基因激活剂LPS(50 ng/ml)和PGN(20µg/ml)诱导RAW264.7和gRNA3组细胞8 h后,荧光定量PCR和ELISA检测TNF-α表达与分泌水平,结果显示,与诱导后正常RAW264.7细胞比较,敲除MyD88基因后TNF-α表达和分泌水平均下调(P<0.05,图5、6)。
3 讨论
基因编辑技术是一种人为对目的基因片段进行基因敲除、特异突变引入或定点转基因修饰的技术[6]。CRISPR/Cas9技术因高效、特异、操作简易、周期短等优点,逐渐成为目前基因编辑的主流,在生物学领域取得广泛进展,并于2020年被授予诺贝尔化学奖[7]。CRISPR/Cas9系统通过sgRNA引导Cas9蛋白结合于基因外显子区,Cas9蛋白对DNA双链进行切割,通过非同源性末端连接导致随机碱基插入/缺失,导致基因发生移码突变,达到基因敲除目的[8]。本研究首先采用慢病毒感染方法获得稳定表达Cas9的RAW264.7细胞,构建及合成MyD88基因的sgRNA,采用慢病毒感染方式将sgRNA导入稳定表达Cas9的RAW264.7细胞,最终敲除RAW264.7细胞中的MyD88基因。
TLRs属于膜型模式识别受体(pattern recognition receptors,PRRs),是单个跨膜非催化性蛋白,主要表达于单核-巨噬细胞、NK细胞、B淋巴细胞及树突状细胞等,由富含亮氨酸重复单位的胞外结构域、跨膜结构域和胞内Toll-白介素1受体(Toll-interleukin 1 receptor,TIR)结构域组成,而TIR的功能是募集下游接头分子转导信号[9]。MyD88蛋白是第一个被发现的TLRs接头分子,其结构也为三部分:N端的死亡结构域(death domain,DD)、中间区域及C端的Toll区[10]。NF-κB是MyD88下游的关键转录因子,通常以p65和p50异二聚体形式存在,细胞静息状态下由于抑制蛋白IKβ结合,致使NF-κB滞留于细胞质,无法调控相关基因转录[11]。当TLRs与配体结合时,TIR与MyD88的C端Toll区形成复合体,引起DD结构域活化,募集IRAK1、IRAK4和TRAF6,TRAF6活化并激活TAK-1,使IKK(IKβkinase)磷酸化。磷酸化的IKK可催化IKβ泛素化降解,解除其对NF-κB的抑制,NF-κB由胞质进入胞核作用于反应原件,启动一系列特定基因转录表达,产生细胞因子TNF-α、IL-1和IL-6等[12]。ADACHI等[13]对MyD88基因敲除的小鼠腹腔注射高剂量LPS,小鼠可存活96 h以上。本研究构建的稳定敲除MyD88基因的RAW264.7细胞株经LPS和PGN诱导后,TNF-α表达明显受抑制,与既往研究结果一致。
最新研究表明,MyD88在固有免疫和适应性免疫中均发挥作用,对自身免疫疾病、炎症性疾病、感染性疾病和过敏性疾病等均有调控效应,但机制尚需进一步探究[14]。本研究采用CRISPR/Cas9系统,借助慢病毒感染方法,成功构建RAW264.7细胞MyD88基因缺失株,可为深入探寻MyD88基因作用提供重要载体。
