催化裂化柴油馏分在离子液体中扩散行为的分子动力学模拟
2022-10-05华渠成王春璐管翠诗
倪 清,华渠成,任 强,龙 军,范 曦,王春璐,管翠诗
(中国石化石油化工科学研究院,北京 100083)
催化裂化柴油(简称柴油)馏分中不同结构类型的烃类组分有不同的用途[1]。例如:链烷烃、环烷烃的十六烷值较高,可作为生产清洁柴油的调合组分,也可作为易裂解组分用于生产高附加值烯烃[2-4];单环芳烃可用于生产基础化工原料苯、甲苯、二甲苯(BTX)产品[5];双环及双环以上芳烃可作为生产碳材料的原料[6-7]。因此,分类富集柴油馏分中的不同烃类组分,获得满足不同加工工艺要求的原料分子成为该领域的研究热点。溶剂抽提技术能够在不改变柴油馏分分子结构的前提下,利用不同烃分子在抽提溶剂中的溶解度差异,分离不同的烃类组分[8]。因此,溶剂抽提技术受到广泛关注。溶剂抽提技术的关键在于抽提溶剂的选择与开发。开发高性能抽提溶剂,提高其溶解目标组分的选择性,降低抽提过程能耗,是溶剂抽提技术的发展方向[9]。
离子液体是近年来兴起的新型溶剂,通过设计阴阳离子结构可以有针对性地调节其对溶质的溶解能力[10-11],因而以离子液体为溶剂分离不同烃类组分的报道日渐增多[12]。与传统有机溶剂相比,离子液体对C6~C14芳烃表现出更高的选择性溶解能力[13],而这种选择性溶解能力可能是由阳离子-芳烃分子间的π—π作用、C—H—π键作用、阴离子-芳烃分子间的氢键作用等形成的[14-15]。为了探讨烃分子与离子液体分子间的相互作用,深入认知溶剂对烃组分选择性溶解能力的形成机理,需要采用分子动力学模拟方法,从分子间作用力、分子形态等微观角度研究溶质分子在离子液体体系中的扩散行为,进而获知烃分子在离子液体中的溶解过程。
本课题选取柴油馏分模型化合物和离子液体1-己基-3甲基咪唑四氟硼酸盐([C6mim][BF4])构建烃分子/离子液体二元共混模型,进行分子动力学模拟,分析烃分子在离子液体体系中的均方位移、烃分子与离子液体分子间的相互作用、烃分子的分子尺寸对共混体系自由体积分数的影响,为新型高选择性离子液体溶剂的开发提供理论参考。
1 模拟体系的建立
1.1 柴油馏分模型化合物的选择
柴油馏分中的烃类组分主要有链烷烃、环烷烃和芳烃。其中,链烷烃碳数分布主要为C11~C22,且以C14~C17最多,以正构烷烃和单取代异构烷烃为主,多取代异构烷烃较少;环烷烃碳数分布一般在C11~C22之间,以C12~C15最多,以单环环烷烃为主;芳烃以双环芳烃为主,单环芳烃和三环及三环以上芳烃相对较少[16]。芳烃中,单环芳烃以烷基苯类为主,碳数分布主要为C10~C15,芳环上烷基侧链的碳数一般为C4~C8;双环芳烃以萘类为主,碳数分布主要为C11~C16,一般含有1~2个侧链,侧链以正构烷基为主;三环及三环以上芳烃以菲类为主,碳数分布主要为C14~C18,芳环烷基侧链的碳数不大于3,以甲基为主[17-18]。
基于上述分析,采用Materials Studio软件构建柴油馏分模型化合物的分子模型,如表1所示。
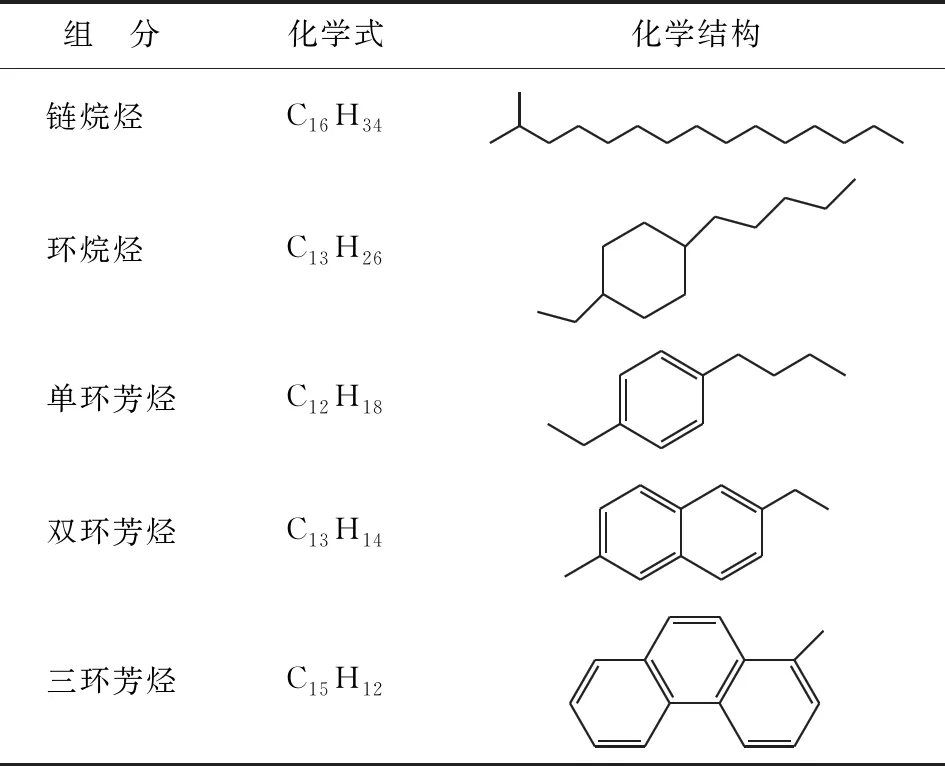
表1 柴油馏分模型化合物及其分子结构
中国石化青岛炼化分公司(简称青岛炼化)和中国石化北京燕山分公司(简称燕山石化)生产的两种柴油的基本物性[19-20]如表2所示。根据两种柴油样品中不同烃类组分的组成,利用Amorphous Cell Tools模块下Construction工具,采用分子动力学方法分别构建两种油样的模拟体系。其模拟参数为:常压、298 K、NPT系综、COMPASS Ⅱ力场;静电交互作用和范德华交互作用分别采用Ewald和Atom Based算法,截断距离为1.25 nm。采用Nose方法控制温度,Berendsen方法控制压力,步长为1 fs,持续时间500 ps。
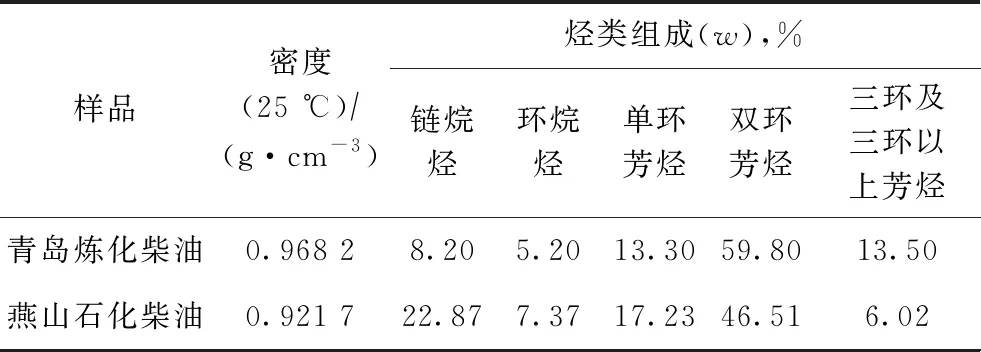
表2 常压条件下两种油样的基本物性
模拟体系平衡状态下不同烃分子的分布,如图1所示。利用Forcite模块中的Density功能统计模拟体系在平衡状态下不同时刻的密度,然后对所有获得的密度求平均,以此平均值作为模拟体系的模拟密度。青岛炼化柴油和燕山石化柴油模拟体系的密度分别为0.959 0 g/cm3和0.921 7 g/cm3,与实际密度(表2)的绝对误差分别为0.95%、2.57%。模拟体系密度计算值与柴油实际密度测量值相符,说明选择的模型物分子结构合理,构建模拟体系的方法较为准确[21]。
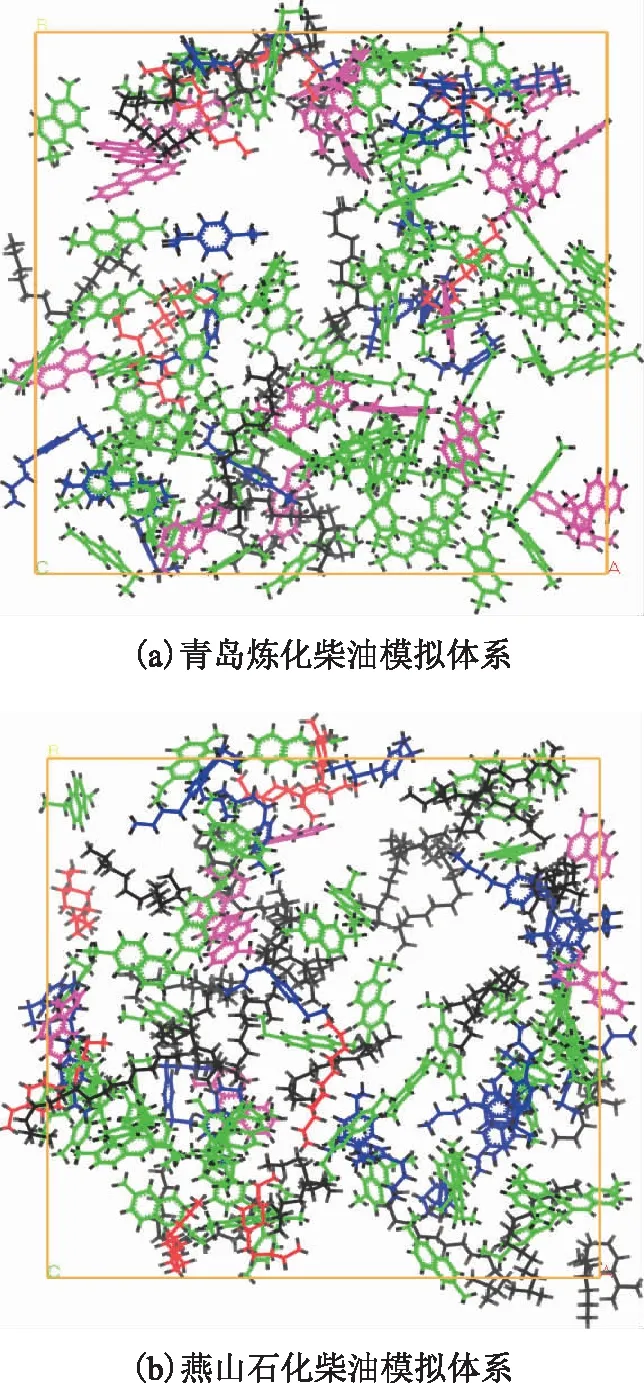
图1 催化裂化柴油馏分模拟体系
1.2 离子液体模拟体系的建立与验证
离子液体[C6mim][BF4]由烷基取代的咪唑阳离子[C6mim]+和四氟硼酸阴离子[BF4]-组成,一般以离子对形式存在[14,22],整体呈电中性。离子液体阴阳离子间的相互作用主要包括静电作用、范德华作用、氢键作用等,其中静电作用的贡献最大。
采用Materials Studio软件构建离子液体[C6mim] [BF4]离子对模型,并在B3LYP/DNP水平下对其进行结构优化和频率分析[23]。结构优化后,无虚频的离子液体离子对构型及其静电势(E)分布如图2所示。由图2可知,F原子与H原子的间距为0.18~0.25 nm,具备形成氢键的条件。负电势集中在阴离子区域,正电势集中在阳离子区域,离子对表面静电势明显高于中性烃分子,说明其具有较强的正/负电荷中心。
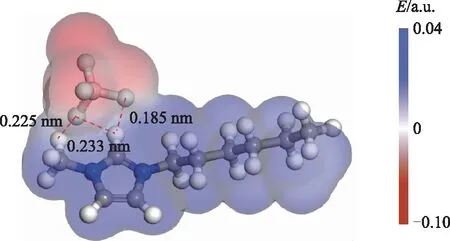
图2 离子液体[C6mim][BF4]的优化构象及表面静电势
模拟体系的准确程度与体系设定的粒子数多少有关。体系粒子数过少,模拟结果波动大,性质不稳定,无法反映真实体系[24]。本研究中,构建离子对数为256的离子液体[C6mim][BF4]模拟体系,并通过对比不同温度下体系密度的模拟值与试验值[25-26]验证所建体系的可靠性。首先,在COMPASS Ⅱ力场下对模拟体系进行200 ps的NVT系综动力学模拟,再进行退火处理(从200 K升温到800 K后再降温到200 K,循环5次);经退火处理后,对模拟体系进行500 ps的NPT系综动力学模拟,时间步长为1 fs,获得平衡态的[C6mim][BF4]模拟体系。
不同温度下离子液体体系密度的模拟值与试验值如图3所示。由图3可知,随着温度升高,离子液体体系密度的模拟值与试验值均呈不断降低的趋势,且二者误差基本在可接受范围内,说明构建离子液体模拟体系的方法可靠。因此,以下将基于该离子对数为256的[C6mim][BF4]体系,讨论烃分子在离子液体中的扩散行为。
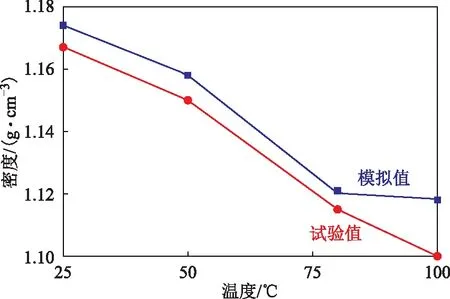
图3 不同温度下[C6mim][BF4]体系密度的模拟值与试验值
2 烃组分与离子液体相互作用的模拟结果
2.1 烃分子在离子液体中的均方位移(MSD)
分子在体系中的MSD可以反映平衡状态下分子运动的剧烈程度。分子的MSD越大,说明分子在该体系中的扩散迁移能力越强[27]。在分子动力学模拟中,分子的MSD是指分子的连续位移随时间的变化关系,可通过式(1)计算[28]。
(1)
式中:MSD为分子均方位移,m2;N为扩散粒子数目;t为扩散时间,s;ri(t)为t时刻的位移,m。
(1)系统参数初始化.确定系统的运行周期t>Tsimulation,初始化窗口内卫星个数N=0,时间t=0;
以所建含有256个离子对的离子液体体系分别与含有128个分子烃组分体系(链烷烃、环烷烃、芳烃等),构建离子液体与不同烃组分的二元共混模型。设定体系温度为298 K,利用Forcite模块讨论烃分子在离子液体体系中的扩散行为。图4为不同烃分子在离子液体中的MSD随着模拟时间延长的变化曲线。
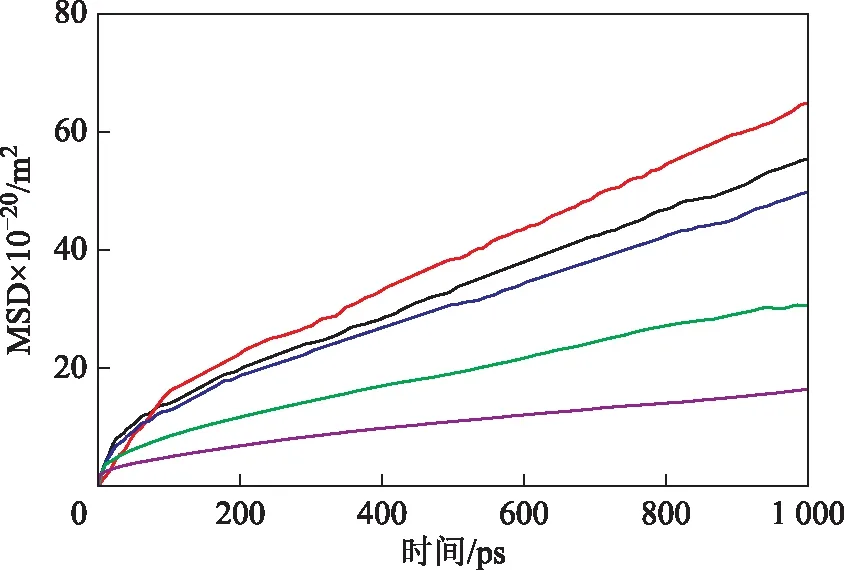
图4 298 K下不同烃分子在离子液体[C6mim][BF4]中的MSD
由图4可以看出,体系达到模拟平衡后,随着模拟时间的延长,不同种类烃分子的MSD均呈线性增大趋势;同一时刻环烷烃的MSD最大、链烷烃的次之、芳烃的MSD较小,而且随着芳烃分子中芳环数增加,其MSD逐渐减小,说明分子的扩散迁移能力随着芳环数的增加而减弱。这是因为离子液体体系是带有正/负电荷中心的强极性体系,易与极性组分发生强相互作用;链烷烃和环烷烃分子的极性较弱,与离子液体的相互作用较弱;芳烃分子中有离域π电子,芳环平面两侧为负电集中区域,分子呈现出一定极性[14],因而芳烃分子与离子液体的作用更强;随着芳环数增加,离域π电子密度增加,芳烃分子极性增强,其与离子液体的相互作用增强,对其扩散位移的限制性更强。
2.2 烃分子与离子对的相互作用
不同化学结构的烃分子与离子液体的作用类型与强弱不同,导致烃分子在离子液体体系中的扩散行为不同。因此,研究烃分子与离子液体间作用的类型和强弱有利于深入认识烃分子在离子液体体系中的扩散机理。
对处于平衡状态的共混体系,不同结构的烃分子与离子液体间的相互作用能可按照式(2)通过Forcite 模块分析[29-30]获得。
Einter=Etotal-E离子液体-E烃分子
(2)
式中:Einter为共混体系中离子液体与烃分子的分子间相互作用能,kJ/mol;Etotal为离子液体与烃分子共同存在时体系分子间的总相互作用能,kJ/mol;E离子液体、E烃分子分别为体系中离子液体、烃同类分子间的相互作用能,kJ/mol。
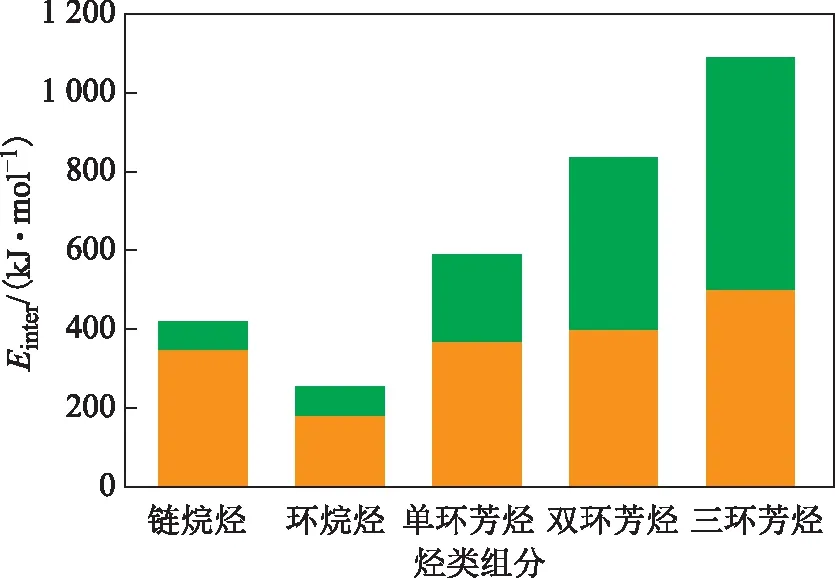
图5 不同烃分子与离子液体[C6mim][BF4]分子间的相互作用能
2.3 烃分子体积与共混体系自由体积的关系
离子液体阴阳离子间具有特殊的氢键作用,使离子液体内部存在广泛的空间网格结构[31]。尺寸较小的溶质分子更容易进入离子液体空间网格体系,与离子液体发生相互作用。因此,探讨柴油馏分模型化合物的分子体积大小,可深入认识烃分子在离子液体体系中扩散的难易程度。
描述分子体积常见的模型有球形模型、圆片/圆柱模型、矩形盒子模型等[32]。由于柴油馏分模型化合物的分子构型更接近于矩形盒子,尤其是多环芳烃分子中具有较大的平面刚性结构,因此选择矩形盒子模型计算烃分子的体积。将优化后无虚频的烃分子结构导入Multiwfn程序[33],图6为采用Corey-Pauling-Koltun模型显示的不同烃分子在能量最低构象下的三视图。由图6可知,不同烃分子的构象较复杂,均为非对称结构,采用矩形盒子模型能够较好地匹配烃分子形状和尺寸。

图6 柴油馏分模型化合物分子的最低能量构象三视图
在共混模型体系中,未被物质占据空间的体积称为自由体积,被离子液体及烃分子占据的空间称为被占据体积。体系的自由体积分数(FFV)为自由体积与总体积之比,可通过式(3)计算[28]。FFV越大,意味着体系越松散,为烃分子提供的扩散空间越大,烃分子可以在体系中剧烈运动。
(3)
式中:FFV为自由体积分数,%;VFree为体系的自由体积,nm3;VOccupied为体系被占据的体积,nm3;
使用Materials Studio中的Atom Volume & Surface工具获得共混体系平衡状态下的自由体积和被占据体积,由式(3)求得共混体系的FFV,结果如表3所示。由表3可知:链烷烃的分子体积最大,其次是环烷烃,芳烃的分子体积最小;芳烃中,随着分子中芳环数的增加,分子体积不断减小;环烷烃与离子液体共混体系的FFV最大,其次是链烷烃与离子液体的共混体系,芳烃与离子液体共混体系的FFV最小,且随着分子中芳环数的增加FFV逐渐减小。
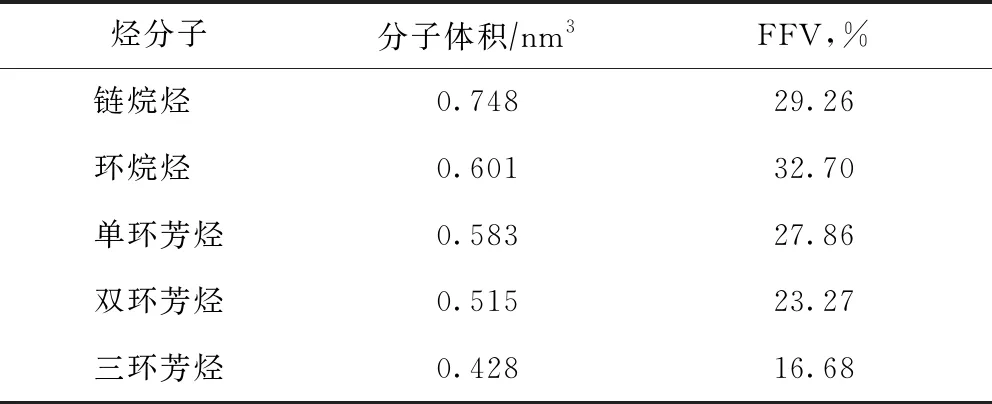
表3 烃分子的三维尺寸及对应共混体系的自由体积分数
这是因为共混体系的FFV与分子间相互作用和分子体积有明显相关性:环烷烃与离子液体的相互作用最弱,其分子在离子液体体系内受到的束缚最小,分子运动最剧烈,导致环烷烃与离子液体共混体系的FFV最大;链烷烃与离子液体的相互作用较弱,分子扩散迁移速率较高;芳烃分子与离子液体分子的相互作用较强,芳烃分子的运动受到二者相互作用的限制,使芳烃分子占据更多自由体积,导致共混体系的FFV降低。而且,随着分子中芳环数增加,芳烃分子的体积逐渐减小,与离子液体的相互作用逐渐增强,单位自由体积容纳的芳烃分子更多,共混体系的FFV不断减小。
图7直观地展现了离子液体与不同烃分子共混体系的自由体积空间分布状态,其中蓝色区域为共混体系的自由体积。由图7可知:离子液体体系存在较多的空隙;由于分子热运动,共混体系中的烃分子会填充离子液体分子间的空隙;随着烃分子体积的减小,共混体系的自由体积呈减小趋势。

图7 不同共混体系自由体积的空间分布状态
3 结 论
(1)对于催化裂化柴油馏分模型化合物分子,饱和烃比芳烃分子更容易在离子液体体系中进行扩散。不同烃分子在离子液体体系中的扩散能力由大到小的顺序为:环烷烃>链烷烃>单环芳烃>双环芳烃>三环芳烃。
(2)烃分子与离子液体的分子间相互作用随着烃分子结构变化而不同。范德华作用是饱和烃分子与离子液体间主要的相互作用。随着芳烃环数增多,芳烃分子与离子液体的静电作用逐渐增强,对于三环芳烃,静电作用是其与离子液体间的主要相互作用。
(3)烃分子在离子液体体系中的扩散行为与烃分子体积及其与离子液体的分子间相互作用有关。体积越小、与离子液体相互作用越强的烃分子在离子液体体系中越不易扩散,其与离子液体形成的共混体系自由体积分数越小。
