基于WGCNA与GSEA方法挖掘调控中卫山羊羊毛弯曲相关基因
2022-09-30李晓波刘占发马月辉赵倩君叶绍辉
李晓波,刘占发,刘 悦,陈 倩,马月辉,赵倩君*,叶绍辉
(1.云南农业大学动物科学技术学院,昆明 650201; 2.中国农业科学院北京畜牧兽医研究所,北京 100193; 3.宁夏回族自治区中卫山羊保种场,宁夏 755000)
羊毛纤维天然卷曲,其弯曲度可以作为评价羊毛品质的依据。中卫山羊是我国珍贵的白色裘皮山羊品种。从1月龄的小羊羔身上取下的毛皮洁白透亮,花纹精美,光泽夺目。羊毛的弯曲表型在生长过程中会逐渐消失,裘皮的经济价值和美观度随着被毛卷曲度的逐渐消失而显著下降。目前,中卫山羊羊毛卷曲随生长发生变化的分子机制尚不明确,需要进一步探索。
毛发生长主要依赖于毛囊结构干细胞的发育,因此毛发形态变化取决于毛囊结构生长发育的状态,毛囊是产生毛发的微型器官,是决定羊毛生长的关键。因此,研究毛囊形态发生的关键基因、信号转导途径和调控机制具有重要意义。毛囊是由胚胎时期的上皮细胞和真皮间充质干细胞及其衍生物通过信号传递相互作用形成的。成熟的毛囊包括真皮乳头、外根鞘、内根鞘、毛球、毛干等结构。毛发表型的形成受到诸多因素影响,细胞层面的研究认为毛发弯曲的主要原因是毛囊细胞的不对称分裂,不对称的细胞分裂和细胞硬化对毛囊底层施加的不同压力导致毛纤维弯曲。在分子方面研究发现,71和基因都在内根鞘(IRS)编码蛋白质,内根鞘是毛囊中的一种刚性结构,通过诱导毛发的生长影响毛发的弯曲度。Dickkopf-1(DKK1)是典型的Wnt信号通路的抑制剂,调节毛囊的形态发生和周期,对1的多态性和与羊毛表型性状的关联分析发现绵羊1基因组区域有11个单核苷酸多态性(single nucleotide polymorphism,SNPs),其中9个与羊毛弯曲有关。通过对胎儿和羔羊皮肤样本的mRNAs表达谱进行分析,有研究发现了一些与毛囊生长发育相关的关键通路,如WNT、TNF、MAPK信号通路。
既往被毛弯曲转录组调控研究主要基于差异基因的GO与KEGG功能富集分析,继而在调控通路中挖掘关键基因。本研究基于WGCNA与GSEA的算法对基因测序数据进行处理从而更有目的性的聚焦于关键的核心基因,对其调控功能进行研究。加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)在挖掘与重要性状及疾病发生发展等生物学过程相关联的核心基因中已经被广泛应用,相比于传统算法,它是一种构建基因共表达网络的系统生物学算法,处理基因的方式更具生物学意义,该方法通过计算软阈值对基因进行加权构建无尺度网络与构建表达矩阵对表达模式相似的基因进行分类,基因被划分到不同的表达模块中通过挖掘模块中发挥核心作用的基因,更加高效准确地为生物学研究提供靶标基因。基因集的富集分析(gene set enrichment analysis,GSEA)是针对基因集合而不是单个基因,它可兼顾差异较小的基因而对所有的差异基因进行富集,通过对基因集进行分析找到与表性差异相关的基因得到的分析结果更可靠。本研究主要应用这两种方法对不同时期的中卫山羊皮肤组织的转录组图谱进行全面的分析。
关于动物毛发弯曲变化相关的基因及调控分子机制的研究报道较少,目前关于中卫山羊羊毛弯曲表型变化的分子机制尚不清楚。本研究旨在构建不同弯曲表型的中卫山羊皮肤组织mRNAs表达谱,鉴定参与毛囊细胞生长发育过程的差异表达的mRNAs,综合利用WGCNA与GSEA分析方法找到调控毛发弯曲的关键基因,本研究为羊毛弯曲的分子机制解析提供新依据及新方法。
1 材料与方法
1.1 试验材料
试验选用的中卫山羊由宁夏回族自治区中卫市中卫山羊保种场提供。在整个试验期间,山羊的饲养条件保持一致。随机选择3只健康、无亲缘关系的羔羊进行活体取样,对同一个体在不同时期进行两次采样,采样位置为轴对称。分别采集45日龄(D45)和365日龄(D365)羔羊皮肤组织,样品保存在RNAlater中用于后续测序,每次采样后均对羊进行抗感染治疗。
1.2 皮肤组织RNA的提取与测序
将保存在RNAlater试剂中的样品提取总RNA进行测序(RNA-seq)。根据试剂盒的说明,使用RNasy Plus Universal Mini Kit(QIAGEN,德国)提取6个样本 (45日龄3个个体,365日龄3个个体)的总RNA。用NanoDrop 2000分光光度计(Thermo Science,Wilmington,DE,美国)初步测定RNA纯度,用Aglient 2100(Agilent Technologies,CA,USA)准确定量RNA样品的浓度。用符合标准的RNA(RNA量>2 μg,浓度 ≥100 ng·μL, RIN ≥7, OD/OD=1.8~2.2, OD/ OD=1.8~2.2) 构建文库。基于Illumina HiSeq 2500平台进行测序。
1.3 不同时期皮肤组织转录组数据分析
利用fastqc 对原始数据进行质控过滤得到clean data,并计算Q20、Q30和GC含量。通过软件TopHat2(v2.2.1),将clean data与山羊参考基因组(版本:assembly ARS1)进行比对。TopHat2可以鉴定外显子之间的剪接位点,将RNA-seq的reads数据map到基因组上。使用Cufflinks软件的Cuffquant和Cuffnorm组件,通过Mapped Reads在基因上的位置信息,对转录本和基因的表达水平进行定量,计算FPKM值。最后,使用CuffCompare将同一组的3个个体进行合并,使用Cuffdiff进行对比获得两个时期之间的差异表达基因集。
1.4 差异表达基因功能富集分析
将差异表达基因的阈值设置为_value <0.05 和 |logFoldChange| ≥1,筛选表达差异显著基因,对其进行后续的分析。利用DAVID和KOBAS在线数据库对差异基因进行GO和KEGG富集分析,以<0.05为阈值筛选显著的GO_Term和Pathways。
1.5 加权基因共表达网络分析与Hub基因筛选
应用WGCNA R包(v1.70)分析基因表达数据。构建了所有差异mRNA的加权基因共表达网络,利用动态分割法寻找模块,并根据表达模式将其划分为不同的模块。分析不同模块之间的相关性,并对所有模块进行聚类。将获得的同一模块内的表达模式相同的差异基因进行功能富集。参数设置如下:CutHeight=0.8,minSize=10,其他参数使用默认值。将基因划分为不同模块后,计算模块的特征值分析模块和样本组间的相关性。选择与不同组间相关性高的模块作为目标模块;对目标模块中的基因进行KEGG富集分析。使用在线工具String(https://cn.string-db.org),设置置信度大于0.85基于模块中差异表达基因构建蛋白互作网络 (protein-protein interaction network, PPI)。剔除无关联的基因,利用Cytoscape 构建核心基因互作网络;利用 cytoHubba插件中的MCC算法选择top10的nodes,得到枢纽基因(hub gene)。
1.6 GSEA分析
本研究应用基因集富集分析(GSEA)方法对两组间基因集合进行基因功能富集分析。GSEA是从全体基因的表达矩阵中找出具有协同差异 (concordant differences)的基因集,因此能兼顾差异较小的基因。参数设置如下:选择“C2.KEGG通路基因集”作为背景,置换检验数量选择1 000默认值,样本量较少置换检验类型选择gene_set,基因类型选择使用基因名,由于本研究样本数量为3个以上,设置的基因排序方法为Signal2 Noise,其他参数设置保持为默认值。
2 结 果
2.1 转录组测序数据质量评估
选取中卫山羊羔羊的两个生长时期(D45,D365),每个时期3个样品,共6个中卫山羊肩部皮肤组织样品进行RNA测序。将数据进行质控、比对、计数,每个样品产生超过12G的原始数据,其中Q30数据基本都在90% 以上,表明测序数据满足后续分析的要求。
2.2 差异表达基因的筛选
为了评估转录本的表达水平,对每个样本表达矩阵的FPKM值进行了归一化处理。发现在6个样本中,45日龄羔羊皮肤组织的mRNA的表达量高于356日龄羔羊(图1A)。这可能与羔羊时期组织器官快速的生长发育有关,这一时期体内新陈代谢旺盛,基因的表达相对更为活跃。将-value<0.05,|logFC|≥1作为阈值筛选差异表达mRNA,45日龄为对照组,365日龄为试验组进行比较,共鉴定出上调表达mRNA 812个,下调表达mRNA 440个(图1B)。基于mRNA的FPKM值对两组差异表达基因进行系统聚类分析(图1C),结果表明同一群体中的个体表达模式相似,而不同时期组间的表达模式差异显著。
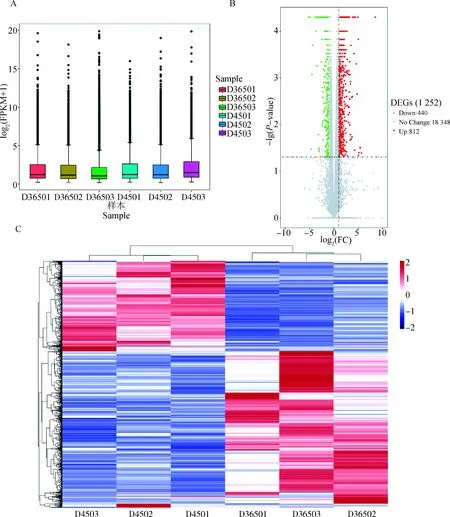
A.个体基因表达量的箱线图;B.差异表达基因的火山图;C.差异基因聚类图A. Box diagram of individual gene expression; B. Volcanic map of differentially expressed genes; C. Cluster map of differential genes图1 不同时期皮肤组织的差异基因的表达、筛选与聚类结果Fig.1 Expression, screening and clustering of differential genes in skin tissues at different stages
2.3 差异表达基因的GO和KEGG富集分析
对1 252个差异基因进行GO和KEGG功能富集分析,共富集到119个GO_term,其中包含86个生物学过程(BP),20个细胞组分(CC),13个生物学功能(MF)。每个项目中前5条被显著富集到的GO条目如图2A所示。在生物学过程中,差异表达基因被显著富集到细胞粘附(cell adhesion,16个基因)、细胞表面受体信号通路(cell surface receptor signaling pathway,12个基因)、吞噬(phagocytosis,6个基因)。在细胞组分当中,差异基因显著富集到细胞外间隙(extracellular space,60个基因)、细胞外囊泡 (extracellular exosome,117个基因)。在生物学功能当中,差异基因显著富集到花生四烯酸结合(arachidonic acid binding,6个基因)和钙离子结合(calcium ion binding,36个基因)。这些富集条目与物质代谢和能量循环过程紧密相关,表明细胞生命活动频繁,毛囊的生长发育受到这些生物学进程的调控。
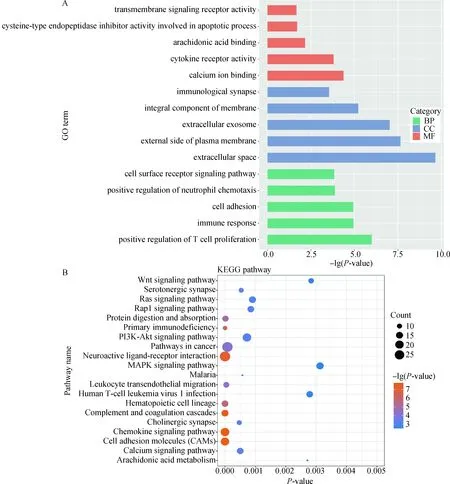
图2 差异基因GO和KEGG富集分析Fig.2 GO and KEGG enrichment analysis of differential genes
KEGG分析结果显示,共79条信号通路被显著富集,图2B列出了前20个显著富集的通路。发现有一些之前的研究中已经被报道参与毛囊发育调控的通路,其中包含PI3K-Akt signaling pathway、Rap1 signaling pathway、Ras signaling pathway、Wnt signaling pathway、MAPK signaling pathway等。
2.4 差异基因的加权共表达网络分析
基于WGCNA方法对D45和D365两组间的1 252个差异基因进行分析。利用 pickSoftThreshold计算软阈值,在R>0.85条件下, 选择最佳软阈值β为22进行无尺度网络的构建(图3A、3B)。根据基因表达特征进行聚类划分模块,共鉴定出14个模块(black、blue、brown、cyan、green、greenyellow、magenta、pink、purple、red、salmon、tan、turquoise、yellow)(表1,图3C),各模块包含基因数量在表1中列出;其中最大的绿松石模块有306个差异基因,浅橙色模块中基因数量最少,仅仅包括7个基因。利用各模块的特征向量值计算模块与两个不同时期羊毛弯曲表型的关系;其中与D45相关性(R=0.988,<0.01)最高的模块为绿松石模块,与D365相关性(R=0.922,<0.01)最高的模块为黄色模块(图4A);将不同模块进行聚类分析,发现绿松石与黄色模块聚集在不同分支。将这两个模块作为目标模块进行后续分析。
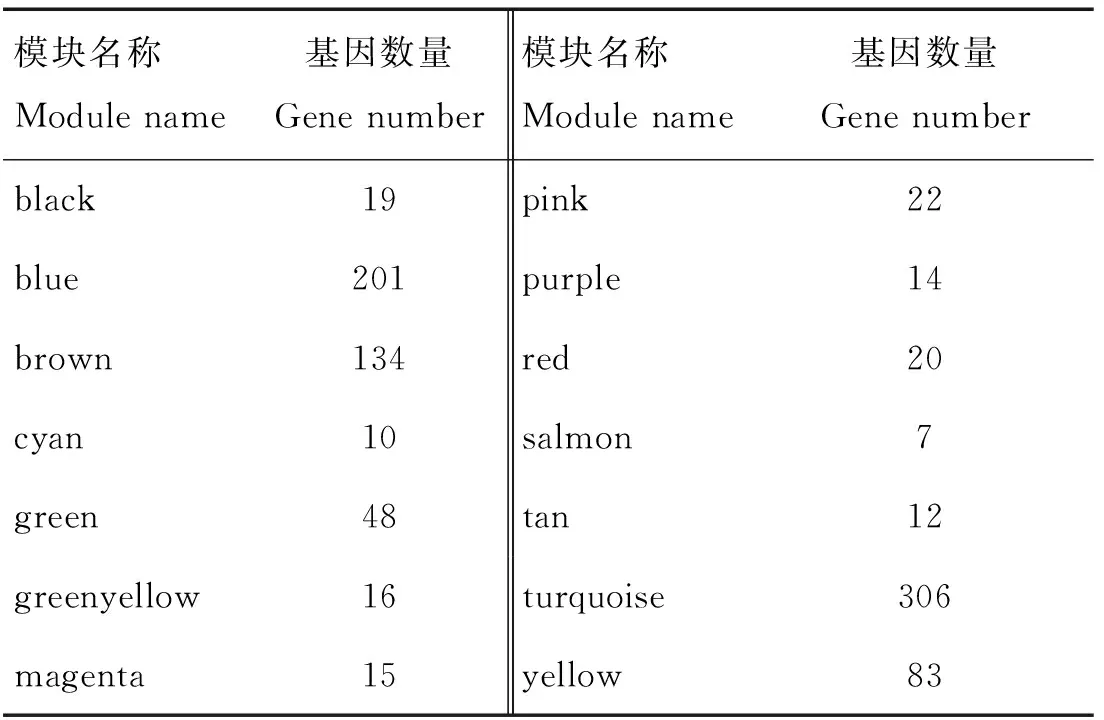
表1 各模块中基因数量Table 1 Number of genes in each module

A.软阈值β计算结果无尺度网络线性模型;B.不同软阈值对应的网络连接度;C.基因层级聚类划分模块A. The result of soft threshold β calculation is a scale-free network linear model; B. Network connectivity corresponding to different soft thresholds; C. Gene hierarchical clustering partition module图3 差异基因加权共表达网络分析Fig.3 Weighted co-expression network analysis of differential genes
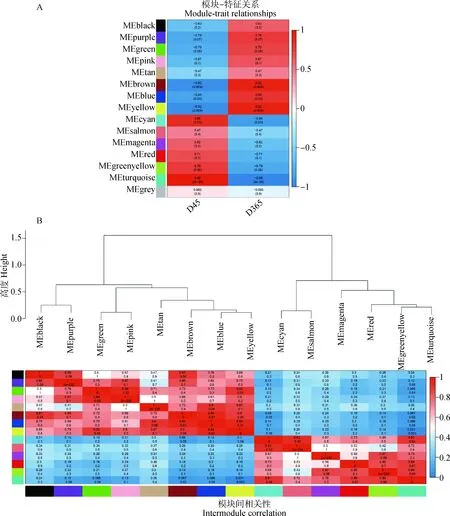
A.模块与表型相关性分析;B.模块相关性分析A. Correlation analysis between modules and phenotypes; B. Module correlation analysis图4 模块相关性Fig.4 Module correlation
2.5 目标模块中基因的KEGG分析
对目标模块中的基因进行KEGG功能分析,结果显示黄色模块中的基因显著富集到JAK/STAT signaling pathway、Histidine metabolism、Chemokine signaling pathway、cGMP-PKG signaling pathway、beta/Alanine metabolism等通路中;绿松石模块中基因富集到Wnt signaling pathway、TGF/beta signaling pathway、Biosynthesis of amino acids等通路中(图5A-B)。其中Jak/STAT、Chemokine、Wnt、TGF/beta通路在调控毛囊发育与毛发表型中被已有的研究多次报道。
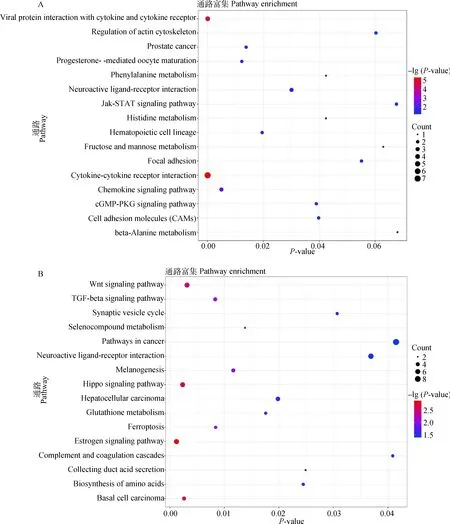
A.黄色模块中基因显著富集通路;B.绿松石模块中基因显著富集通路A. The significant enriched pathways by genes in yellow module; B. The significant enriched pathways by genes in turquoise module图5 目标模块基因的KEGG分析Fig.5 KEGG analysis of target module genes
2.6 目标模块分子网络分析鉴定关键基因
对目标模块中基因进行蛋白质-蛋白质互作网络(protein-protein interaction, PPI)的构建,分析蛋白之间的相互作用。黄色模块中基因获得18个蛋白质节点,绿松石模块中基因获得41个蛋白质节点(图6A-B)。在Cytoscape 中将结果导入构建核心基因互作网络,利用cytoHubba插件中的MCC算法选择top10的nodes。黄色模块中得到的10个核心基因为27、7、20、26、1、10、4、、31、2 (图7A)。绿松石模块中得到的10个核心基因为71、75、14、78、15、1、2、3、、3 (图7B)。

A.黄色模块中基因的PPI网络;B.绿松石模块中基因的PPI网络A. PPI network of genes in yellow module; B. PPI network of genes in turquoise module图6 目标模块基因蛋白互作网络Fig.6 Target module gene protein interaction network
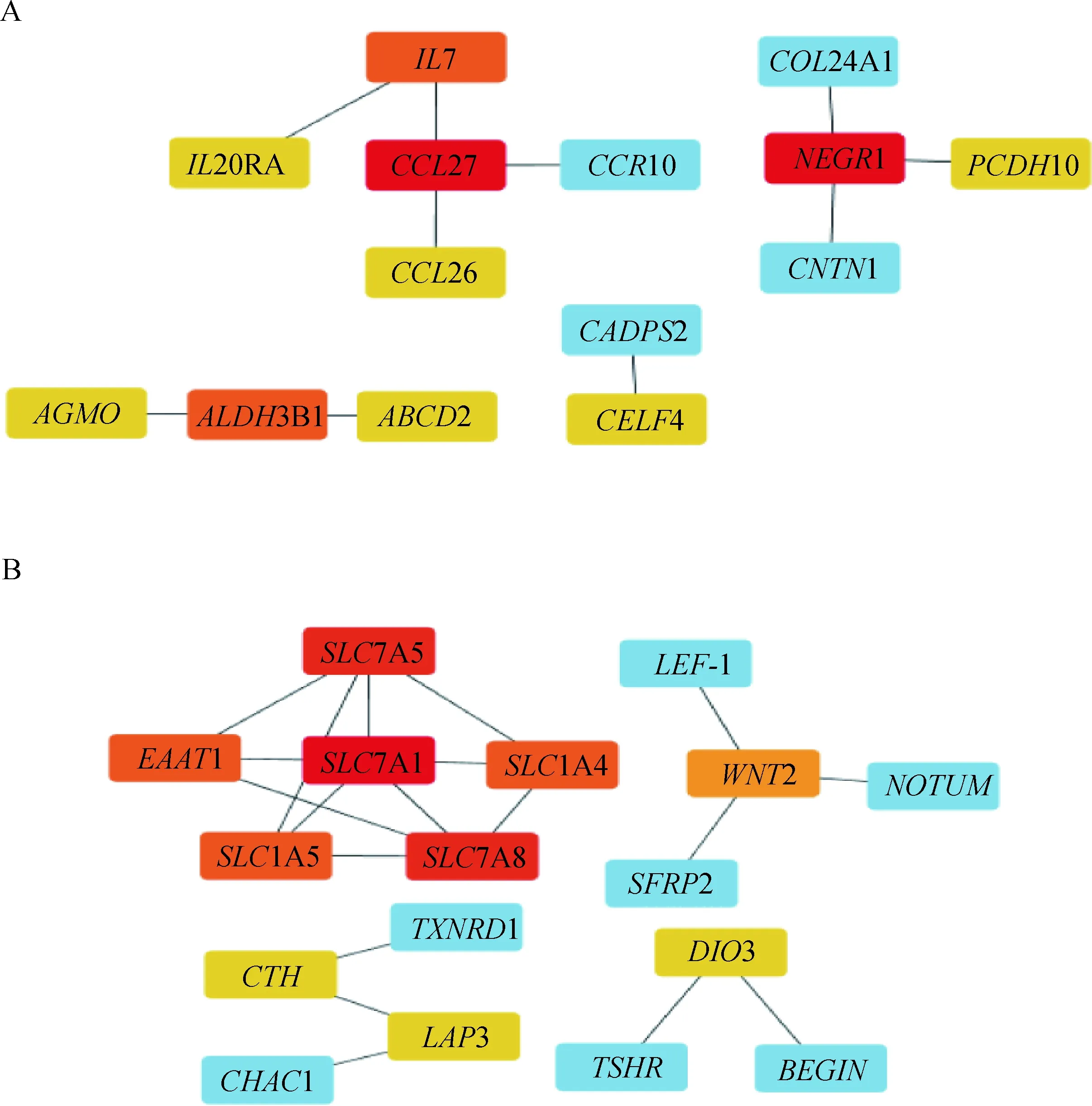
A.黄色模块Top10基因;B.绿松石模块Top10基因。颜色由蓝到红,红色越深说明连接度越高A. Top10 genes in yellow module;B. Top10 genes in turquoise module. The color ranges from blue to red, and the darker the red, the higher the connection图7 目标模块核心基因网络Fig.7 Core genes network of target module
2.7 基因集合富集分析GSEA
利用GSEA方法对两组样本的所有基因集进行分析。D45样本中73条通路中的基因表达上调,75%的正确率支持其中16条通路的基因上调,nominal-value<0.05的有17条通路;nominal-value <0.01的有9条通路,其中包括Hedgehog signaling pathway、cell cycle、Proteasome、RNA degradation、TGF/BETA信号通路等。D365样本中有93条通路中的基因表达上调,75%的正确率支持其中30条通路的基因上调,nominal-value<0.05的有27条通路;nominal-value <0.01的有18条通路,其中包括Cytokine receptor interaction、JAK/STAT signaling pathway、cell adhesion molecules cams等。先前研究显示,Hedgehog signaling pathway、JAK/STAT signaling pathway、TGF/BETA signaling pathway参与毛囊发育调控。Hedgehog signaling pathway中富集到的15个基因在D45中是上调的;JAK/STAT signaling pathway中富集到的56个基因在D365中是上调的,且2、7、26、8、11调控毛囊生长发育的基因被包含在领头亚基中,因而推测这些集合中的基因在调控毛发弯曲表型变化中发挥了作用(图8A、8B)。
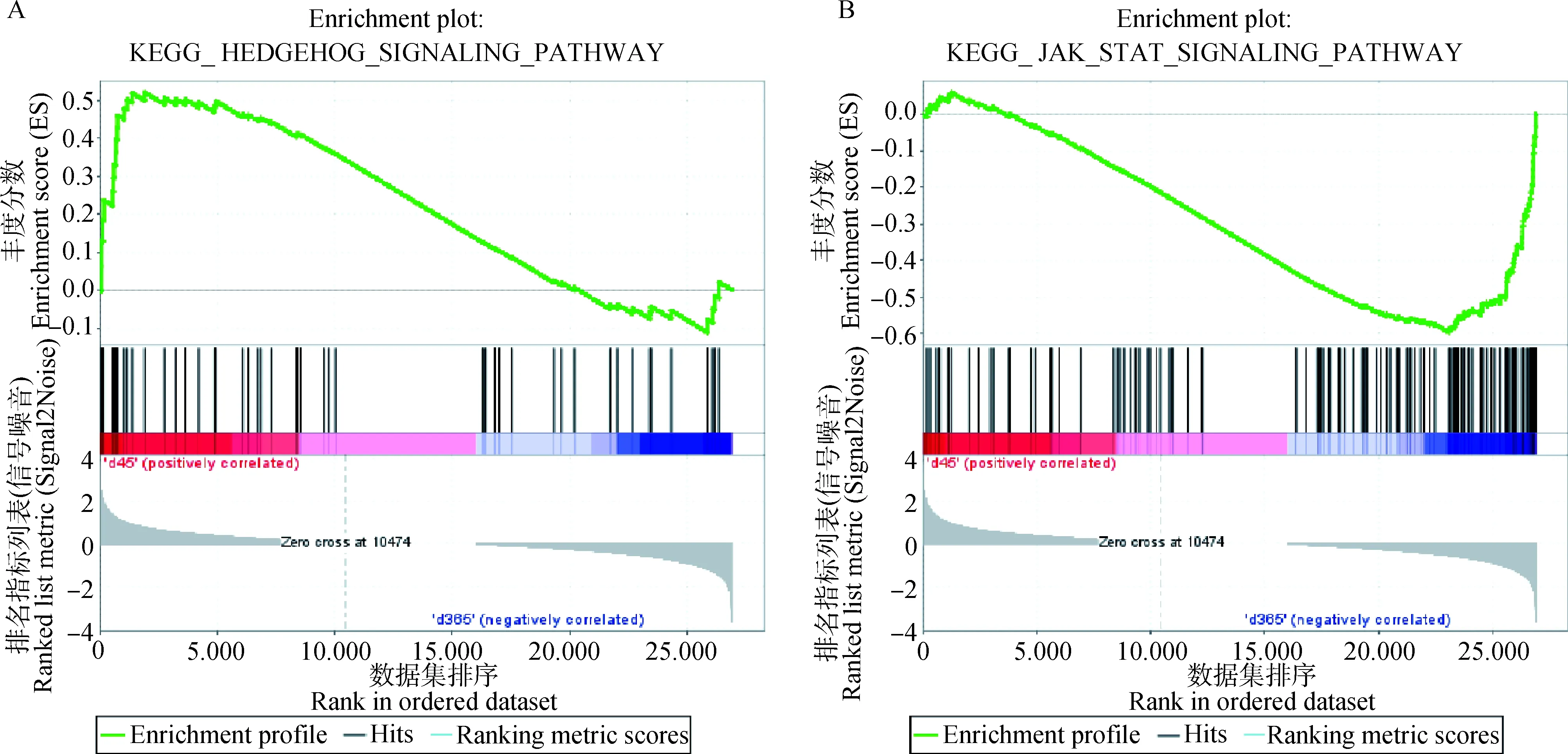
图8 差异基因集合富集分析Hedgehog和JAK/STAT通路结果Fig.8 Results of Hedgehog and JAK/STAT pathways analysis based on set enrichment of differential genes (GSEA) method
3 讨 论
随着高通量技术的发展与应用,畜禽的组学数据不断的充分完善,利用更高级的测序手段和生物信息学手段去解析这些庞大的数据集,更有利于深入地探究畜禽重要性状的分子调控机制。中卫山羊毛曲度随生长发育变化的调节机制尚不明确。为了探讨差异基因对山羊不同生长阶段毛发表型变化的潜在影响,本研究构建了45日龄和365日龄山羊皮肤组织转录组的表达谱,共筛选到1 252个差异显著基因,发现在45日龄皮肤组织中基因总体表达量高于365日龄。本研究综合应用KEGG、WGCNA和GSEA 多种分析方法,对差异表达基因进行功能分析,将富集到参与毛囊发育相关的信号通路和基因集深入解析,进一步筛选影响羊毛弯曲表型的关键Hub基因,阐明中卫山羊毛发弯曲的潜在调控机制。
WGCNA分析可将表达模式高度相关的基因划分为模块,通过计算模块之间的相互关联度以及与外部样本性状的关联,筛选出特异性模块构建分子网络挖掘出关键的核心基因。近年来,WGCNA应用最多的是在研究调控癌症的关键基因,通过模块基因的构建筛选找到致病的Hub基因,在胃癌、肺癌、葡萄球膜黑色素瘤、腹主动脉瘤、乳腺癌等研究中作为主要分析手段。GSEA分析使用预设的基因集,将所有的差异基因根据程度进行排序,然后富集在这个排序的顶端或低端,可筛选到差异表达不显著却有生物学意义的基因。
毛囊细胞生长与形态变化导致羊毛产生弯曲变化。毛囊作为毛发生长的基本器官,在毛发生长发育过程中受到多种信号通路的调控,决定了毛发的表型特征。以往的研究发现,PI3K-Akt、Chemokine、cAMP和RAS和MAPK等信号通路在毛囊发育中起直接或间接作用。有研究发现,表皮细胞迁移分化、毛囊发育和外胚层胎盘形成导致的皮肤结构差异被Wnt和Hedgehog信号通路为代表的KEGG通路调控。本研究对1 252个差异显著的基因进行KEGG分析,结果显示这7条通路均被显著富集。本研究中,WGCNA 根据表达模式将基因划分在14个模块,其中与羊毛弯曲表型密切相关的黄色模块和绿松石模块中基因的KEGG结果显示,JAK/STAT、Chemokine、Wnt、TGF/beta等通路被显著富集。毛发弯曲度的变化可能与这些通路密切相关。通过对目标模块中基因进行分子网络构建,筛选出20个关键调控基因,其中包括27、7、2、1、71等。其中27、7、2与毛囊的生长调控有关,其余基因对毛发弯曲度的调控作用还未见报道。现有的研究发现,皮肤相关趋化因子27调控上皮细胞在控制细胞稳态起重要作用,维持毛囊的生长。7是毛囊中的一种细胞因子,它维持了细胞的动态平衡,对皮肤组织至关重要。作为一个关键因子,2可以调节毛囊的形态发生和发育。推测27、7、2这3个基因在调控中卫山羊毛发弯曲变化中发挥了关键作用。在GSEA富集的通路中,27、7、2分别富集到了Hedgehog signaling pathway和JAK/STAT signaling pathway中。初级纤毛是真核细胞中的细胞器,通过改变信号蛋白的局部浓度和活性来解码各种信号,Hedgehog信号被激活,控制毛发发育的细胞与细胞之间的相互作用。JAK信号转导和转录激活因子STAT信号在控制毛发周期中起着重要作用。根据WGCNA与GSEA分析结果与文献报道推断,27、7、2可能是羊毛产生弯曲的关键基因。
综上所述,本研究构建了中卫山羊不同弯毛表型皮肤的转录组表达图谱。在45和365日龄山羊皮肤组织中共鉴定出1 252个差异表达基因,综合KEGG、WGCNA和GSEA 多种分析方法筛选到了一系列参与毛囊发育和羊毛弯曲度的信号通路和基因。本研究结果可为毛囊发育和羊毛弯曲机理的研究提供依据。
4 结 论
中国中卫山羊优质的羔羊羊皮具有很高的经济价值。羊皮的品质主要受毛发弯曲度的影响,毛囊的生长发育对羊毛的曲度起着重要的调节作用。本研究利用RNA测序技术构建了45和365日龄中卫山羊mRNA的表达谱,综合应用WGCNA和GSEA等生物信息学方法研究了调控毛发弯曲度的相关基因,通过筛选发现Hedgehog、JAK/STAT等信号通路和27、7、2 等基因在调控毛囊发育和羊毛弯曲方面发挥了重要作用。
