检测实验室同时运行多种质量体系的风险与应对措施
2022-09-28刘超武梁燕珍曾国驱
刘超武,梁燕珍,曾国驱,*
1.广东省科学院微生物研究所,广州 510070
2.广东省微生物分析检测中心,广州 510070
3.华南应用微生物国家重点实验室,广州 510070
4.广东省菌种保藏与应用重点实验室,广州 510070
第三方检测实验室为了满足产品认证市场不同质量管理体系的要求,往往需要同时维持包括中国计量认证(China Metrology Accreditation,CMA)、中国国家实验室认可(China National Accreditation Service for Conformity Assessment,CNAS)和良好实验室规范(Good Laboratory Practices,GLP)等各种资质,以此达到不同的质量目标。以公众客户群体满意度为目标的检测实验室,注重检测的技术能力和公正性,遵从CMA和CNAS质量系统[1-2]。另外一类实验室以健康与环境安全评价为目标,注重检测结果和测试全过程,这类实验室以GLP规范作为实验室质量系统[3-4]。
实际上,实验室质量管理体系都是为了保证测试数据的准确、真实和可靠,主要不同在于管理目标。遵从CMA、CNAS采用ISO/IEC 17025质量管理体系,关注产品的质量规范,是产品质量的表现形式。而GLP是一种综合测试与评估手段,用于产品的环境安全和健康风险管理。当监管部门认为ISO/IEC 17025体系不能满足管理要求时,这时需要选择能够最大限度地反映测试过程和结果的管理工具。例如,采用ISO/IEC 17025质量管理体系足以对产品的生物降解性进行认证,而评价降解材料的环境安全和健康风险时,GLP质量管理体系则更适合[5-7]。
GLP质量管理体系由于详尽的测试记录和规范的试验操作,理论上可以复原整个测试过程,因此在事后监管具有明显的优势。ISO/IEC 17025关注结果的有效性,在以测试性能指标为目的时,具有明显的快速筛查能力[5-6]。我们基于检测实验室的管理经验,发现认清不同质量管理体系的特点尤其重要。因此,本文希望通过实例深入剖析不同质量体系管理风险,为检测行业的发展提供参考。
1 质量系统发展历程(Development history of quality systems)
1.1 检测校准质量系统
ISO/IEC 17025体系的建立,首先是国际认可实验室组织(International Laboratory Accreditation Cooperation,ILAC)在1978年起草的检测实验室认可技术准则“检测实验室基本技术要求”,作为国际标准《ISO/IEC指南25:1978实验室技术能力评审指南》第一次发布[8]。之后,经历了第二版《ISO/IEC指南25:1982检测实验室基本技术要求》[9]、第三版《ISO/IEC指南25:1990检测和校准实验室能力的通用要求》[10]。1999年,国际标准化组织(International Organization for Standardization,ISO)和国际电工委员会(International Electrotechnical Commission,IEC)在结合第三版ISO/IEC指南25和ISO 9000系列标准(1994版),联合发布了第一版《ISO/IE 17025:1999检测和校准实验室能力的通用要求》[11]。2005年修订为第二版《ISO/IE 17025:2005检测和校准实验室能力的通用要求》[12]。2017年公布了最新版的ISO/IE 17025的标准[13]。
中国合格评定国家认可委员会的CNAS实验室认证认可工作是在2001年国家认监委成立后正式开展[14],CNAS第一次采用ISO/IE 17025是2006年等同采用第二版《ISO/IE 17025:2005检测和校准实验室能力认可准则》(CNAS-CL01 2006)[15],2018年又结合中国行业特点发布新版认可规范《检测和校准实验室能力认可准则》(CNAS-CL01 2018)[2],以取代旧版文件(CNAS-CL01 2006/ISO 17025 2005)。
中国检验检测机构资质认定始于1985年颁布的《中华人民共和国计量法》,其中规定了对检验机构的考核要求。1987年颁布了《计量法实施细则》对检验机构的考核称为计量认证。之后,检验检测机构资质认定CMA评审准则经历了不断革新,1990年《产品质量检验机构计量认证技术考核规范》(JJF 1021—1990)等同采用ISO/IEC导则25:1990,2000年《产品质量检验机构计量认证/审查认可(验收)评审准则》(试行)等同采用ISO/IEC 17025:1999。2006年《实验室资质认定评审准则》、2016年《检验检测机构资质认定评审准则》与ISO 17025标准更新保持同步。新版CMA(RB/T214—2017)[1,16]对原本19项要素进行了扩充,同时匹配了ISO/IEC 17025:2017最新内容。
CNAS实验室认可是中国与国际接轨的一套国家实验室认可体系,符合国际上通行的校准与检测实验室能力的通用要求。校准检测实验室按照ISO/IEC 17025运行,该标准既是实验室开展有效质量活动的基本要求,也是认证认可机构对实验室能力认可的基本准则。中国计量认证CMA是法律法规规定的强制性行为,其运行模式为国家统一管理,以适应国家法制的需要,其工作注重国际通行做法,并充分考虑中国国情和计量认证实践,是具有中国特点的政府行政认可[7]。虽然CMA评审同样均基于ISO/IEC 17025体系,但CMA较CNAS认可范围更加严格,CMA更多倾向对国家标准的审查,且对国际标准与技术规范通常不予认可。
1.2 GLP质量系统
GLP是产品登记注册非临床健康和环境安全研究的计划、实施、监督、记录、存档和报告的一套质量体系,是政府部门对GLP机构的一种合规性监督[17-19]。GLP始于新西兰,真正实施是1979年美国颁布的第一部药品GLP[20]。1982年,经济合作与发展组织(OECD)颁布实施化学品GLP,明确指出GLP的目的是促进试验数据互认,这使得OECD GLP准则成为国际通行的准则[21]。目前,已有30多个国家和地区签订了协议,表示遵循OECD GLP准则,OECD各成员国通过数据互认协议的共享机制,减少了不必要的重复试验,有效缩短了登记时间[22]。
目前,中国、日本和欧盟等在制定本土GLP法规时参考了OECD GLP准则,英国和德国等只制定了一部通用型的GLP,而美国和日本等国则制定了不同领域的GLP[23-24]。我国于20世纪80年代末和90年代初相继开展了不同产品的GLP工作,目前开展GLP管理的领域主要有食品药品、农药兽药、化妆品和工业化学品GLP实验室评价[6,19,25-28]。
2 ISO/IEC 17025与GLP异同点(Similarities and differences between ISO/IEC 17025 and GLP)
CNAS和CMA覆盖了ISO/IEC 17025的质量、技术和行政要素,核心内容为设备、样品和标准物质、量值溯源、校准、检测方法和质量保证[7],这些要求用来评价实验室校准或检测能力是否达到预期要求,最新标准条款比较如表1所示。

表1 我国国内质量体系最新标准的条款比较Table 1 Comparison of terms among the latest domestic quality system standards in China

续表1CNAS CL01: 2018 (CNAS)RB/T 214: 2017 (CMA)GB/T 22278: 2008 (GLP)7.6测量不确定度的评定 Evaluation of measurement uncertainty4.5.15测量不确定度 Measurement uncertainty/7.7结果有效性的保证 Guarantee of effectiveness of results4.5.19结果有效性 Validity of results/7.8结果的报告 Report of results4.5.20结果报告 Results report4.5.21结果说明 Result description4.5.22抽样结果 Sampling result4.5.23意见和解释 Comments and explanations4.5.24分包结果 Subcontracting result4.5.25传送和格式 Transfer and format4.5.26修改 Modify3.9研究结果的报告 Report of study results7.9投诉 Complaint4.5.8投诉 Complaint/7.10不符合工作 Non conformance4.5.9不符合工作控制 Non conformance control/7.11数据控制和信息管理Data control and information management4.5.16数据信息管理 Data information management/8管理要求 Management requirements4.5管理体系 Management system3.2质量保证计划 Quality assurance programme8.1方式 Options//8.2管理体系文件(方式A) Management system documentation (Option A)4.5.1总则 General4.5.2方针目标 Policy and objectives3.2.1总则 General3.2.2质量保证人员的责任Responsibilities of the quality assurance personnel8.3管理体系文件控制(方式A) Control of management system documents (Option A)4.5.3文件控制 Document control3.10记录存档 Storage and retention of records and materials8.4记录控制(方式A)Control of records (Option A)4.5.11记录控制 Record control4.5.27记录和保存 Records and storage3.10记录存档 Storage and retention of records and materials8.5风险和机会的管理措施 Actions to address risks and opportunities (Option A)4.5.10纠正措施、应对风险和机遇的措施和改进 Measures and improvements to deal with risks and opportunities/8.6改进(方式A) Improvement (Option A)4.5.7服务客户 Serving customers4.5.10纠正措施、应对风险和机遇的措施和改进 Measures and improvements to deal with risks and opportunities/8.7纠正措施(方式A) Corrective actions (Option A)4.5.10纠正措施、应对风险和机遇的措施和改进 Measures and improvements to deal with risks and opportunities/8.8内部审核(方式A) Internal audits (Option A)4.5.12内部审核 Internal audits/8.9管理评审(方式A) Management reviews (Option A)4.5.13管理评审 Management reviews/
以GLP的10个技术要素为参照(表1:3.1~3.10),与ISO/IEC 17025的2个质量体系进行对比分析,ISO/IEC 17025与GLP管理体系的主要异同点如表2所示。
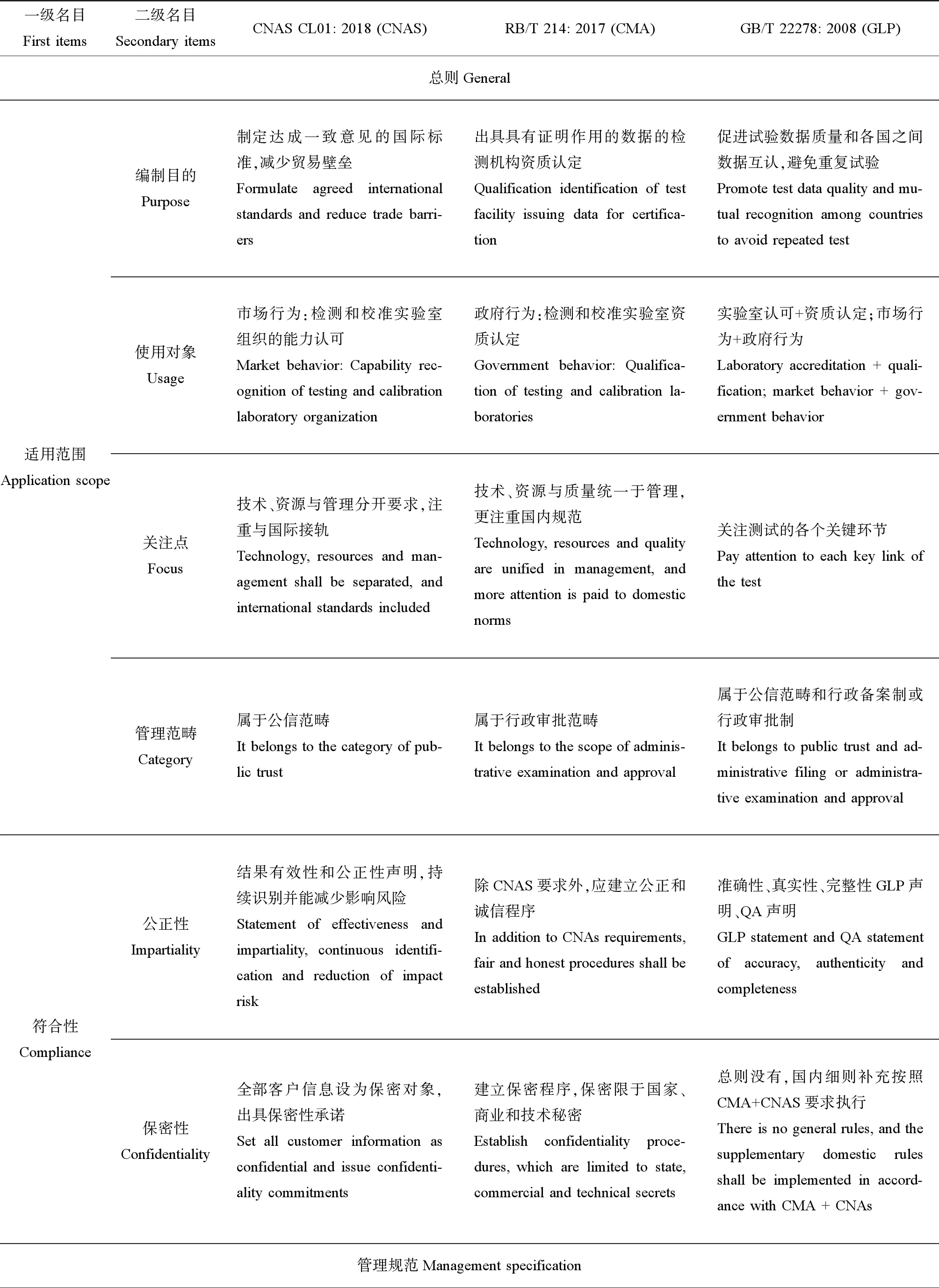
表2 我国国内质量体系最新标准内容比较Table 2 Comparison of the latest domestic quality system standards in China
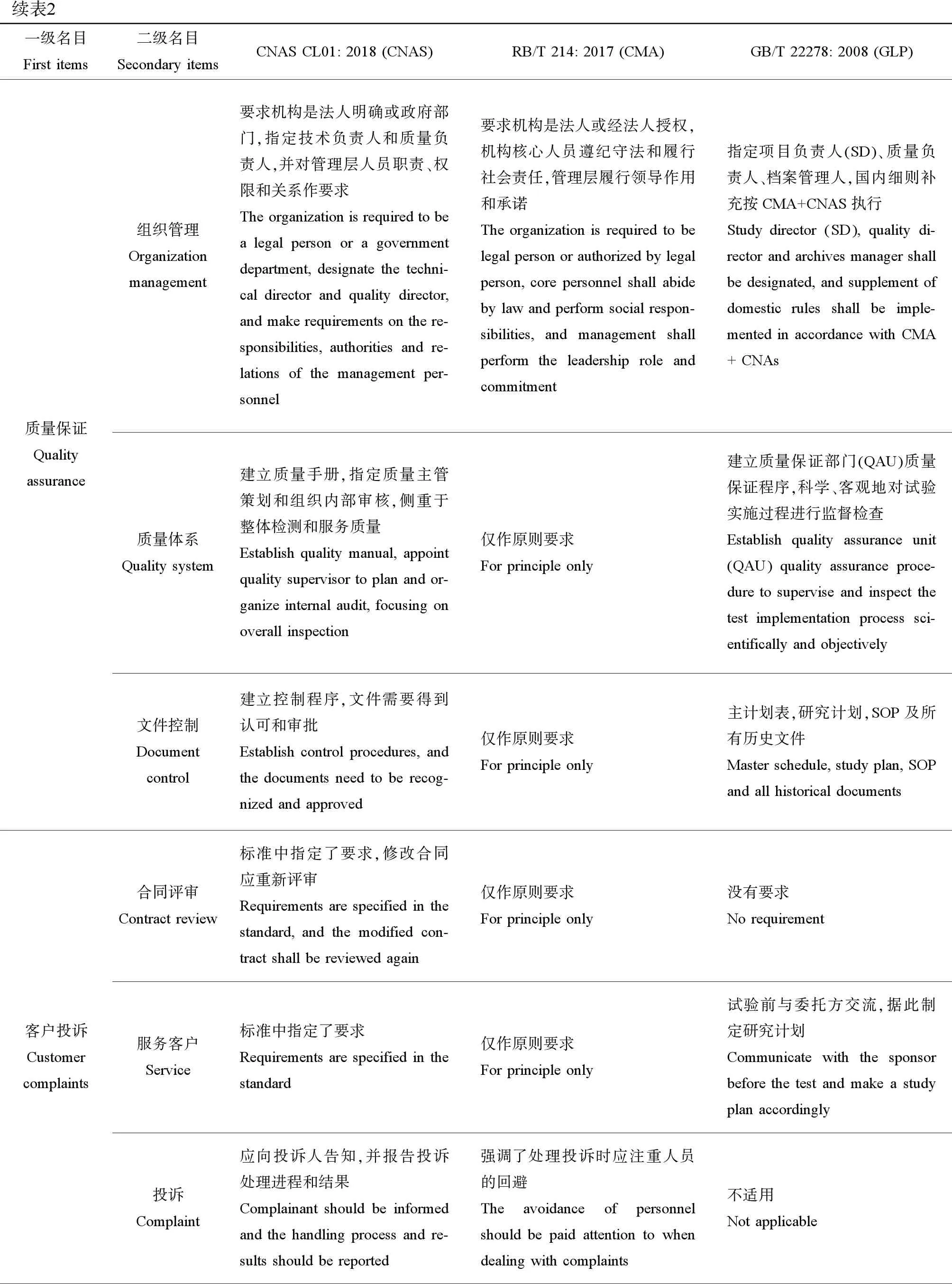
续表2一级名目First items二级名目Secondary itemsCNAS CL01: 2018 (CNAS)RB/T 214: 2017 (CMA)GB/T 22278: 2008 (GLP)质量保证Quality assurance组织管理Organization management要求机构是法人明确或政府部门,指定技术负责人和质量负责人,并对管理层人员职责、权限和关系作要求The organization is required to be a legal person or a government department, designate the techni-cal director and quality director, and make requirements on the re-sponsibilities, authorities and re-lations of the management per-sonnel要求机构是法人或经法人授权,机构核心人员遵纪守法和履行社会责任,管理层履行领导作用和承诺The organization is required to be legal person or authorized by legal person, core personnel shall abide by law and perform social respon-sibilities, and management shall perform the leadership role and commitment指定项目负责人(SD)、质量负责人、档案管理人,国内细则补充按CMA+CNAS执行Study director (SD), quality di-rector and archives manager shall be designated, and supplement of domestic rules shall be imple-mented in accordance with CMA + CNAs质量体系Quality system 建立质量手册,指定质量主管策划和组织内部审核,侧重于整体检测和服务质量Establish quality manual, appoint quality supervisor to plan and or-ganize internal audit, focusing on overall inspection仅作原则要求For principle only建立质量保证部门(QAU)质量保证程序,科学、客观地对试验实施过程进行监督检查Establish quality assurance unit(QAU) quality assurance proce-dure to supervise and inspect the test implementation process sci-entifically and objectively文件控制Document control建立控制程序,文件需要得到认可和审批Establish control procedures, and the documents need to be recog-nized and approved仅作原则要求For principle only主计划表,研究计划,SOP及所有历史文件Master schedule, study plan, SOP and all historical documents客户投诉Customer complaints合同评审Contract review标准中指定了要求,修改合同应重新评审Requirements are specified in the standard, and the modified con-tract shall be reviewed again仅作原则要求For principle only没有要求No requirement服务客户Service标准中指定了要求Requirements are specified in the standard仅作原则要求For principle only试验前与委托方交流,据此制定研究计划Communicate with the sponsor before the test and make a study plan accordingly投诉Complaint应向投诉人告知,并报告投诉处理进程和结果Complainant should be informed and the handling process and re-sults should be reported强调了处理投诉时应注重人员的回避The avoidance of personnel should be paid attention to when dealing with complaints不适用Not applicable

续表2一级名目First items二级名目Secondary itemsCNAS CL01: 2018 (CNAS)RB/T 214: 2017 (CMA)GB/T 22278: 2008 (GLP)实施手段Implementation内部审核Internal audit建议每12个月一次Every 12 months is recommen-ded由质量负责人策划,通常为每年一次Planned by the person in charge of quality; usually once a year质量保证部门有计划的现场检查和定期检查The quality assurance department has planned on-site inspection and regular inspection管理评审Management review由管理层组织负责,通常12个月一次It is organized by the manage-ment, usually once every 12 months由最高管理者负责,通常12个月一次Top management is responsible, u-sually once every 12 months不适用Not applicable外部评审External review通常3年一次,中间进行一次复评审,新项目开展扩项评审Usually once every three years, re-review is conducted in the middle, and the expansion review is carried out for new projects3年一次,中间进行一次复评审,新项目扩项评审Once every three years, re-review is conducted in middle, and ex-pansion review is carried out for new projects各部门并不一致,从3年一次到有因检查不等The departments are not consist-ent, ranging from once every three years to due inspection质量控制Quality control能力验证Capability verification实验室内部和外部开展比对实验、盲样测试、扩项实验Comparison experiment, blind sample test and expansion experi-ment are carried out inside and outside the laboratory仅作原则要求For principle only国内细则补充GLP机构按CMA+CNAS执行GLP facility supplemented by domestic rules shall be imple-mented in accordance with CMA + CNAs质量评估与改进Evaluation and improvement技术人员具备评估偏离的能力,质量评估采用不符合项,指定纠正改进措施相关要求Technicians have the ability to e-valuate deviations and non-con-formities, and specify require-ments for improvement measures仅对不符合项和纠正改进作原则要求 Only principle requirements are made for non-conformities and improvement等同为研究补充和修改;国内细则补充GLP机构按CMA+CNAS执行Equivalent to supplement and modification; GLP facility sup-plemented by domestic rules shall be implemented in accordance with CMA + CNAs风险管理Risk management 指定了预防措施相关要求Requirements for preventive measures are specified仅作原则要求For principle only不适用Not applicable
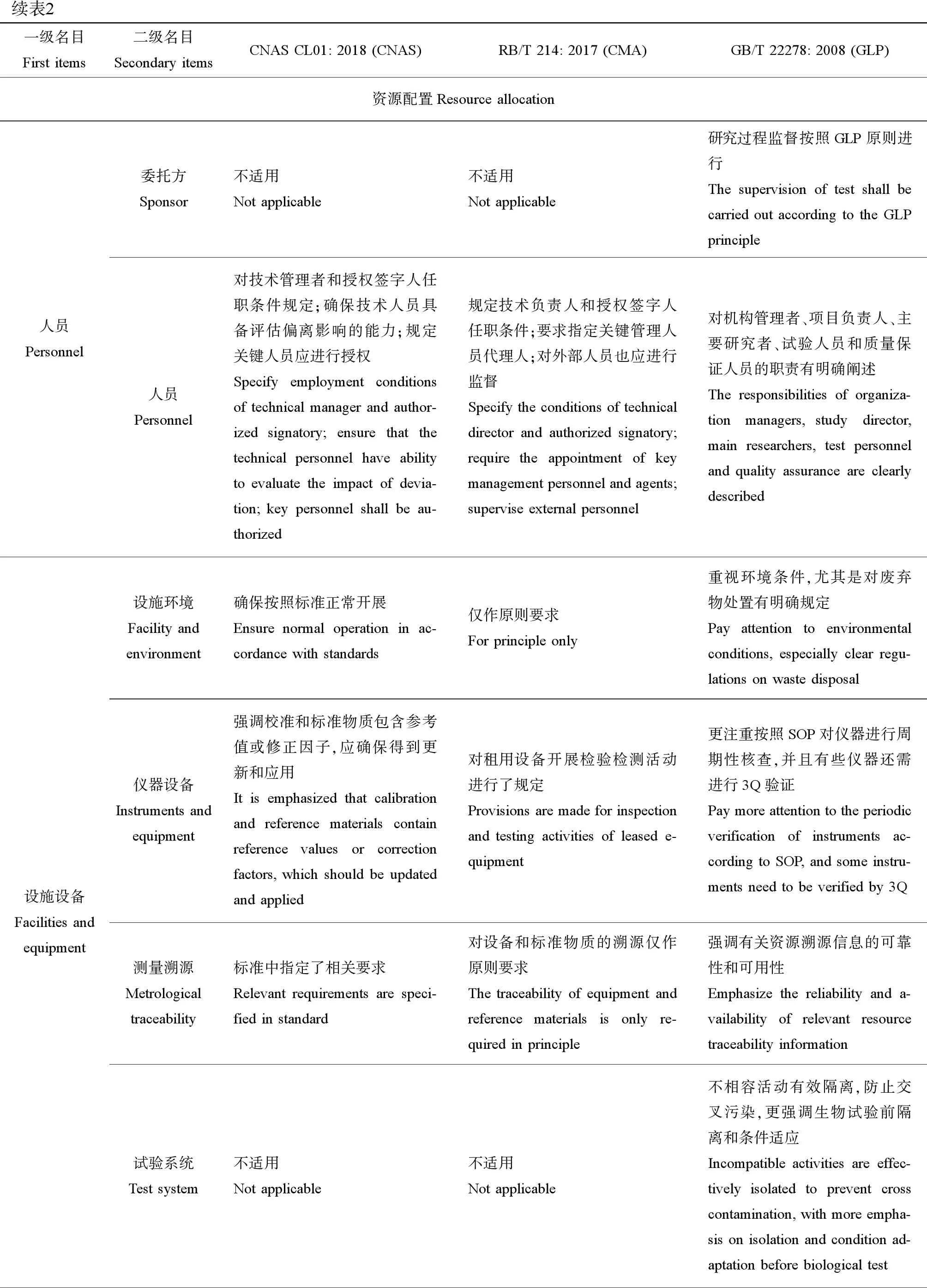
续表2一级名目First items二级名目Secondary itemsCNAS CL01: 2018 (CNAS)RB/T 214: 2017 (CMA)GB/T 22278: 2008 (GLP)资源配置Resource allocation人员Personnel委托方Sponsor不适用Not applicable不适用Not applicable研究过程监督按照GLP原则进行The supervision of test shall be carried out according to the GLP principle人员Personnel对技术管理者和授权签字人任职条件规定;确保技术人员具备评估偏离影响的能力;规定关键人员应进行授权Specify employment conditions of technical manager and author-ized signatory; ensure that the technical personnel have ability to evaluate the impact of devia-tion; key personnel shall be au-thorized规定技术负责人和授权签字人任职条件;要求指定关键管理人员代理人;对外部人员也应进行监督 Specify the conditions of technical director and authorized signatory; require the appointment of key management personnel and agents; supervise external personnel对机构管理者、项目负责人、主要研究者、试验人员和质量保证人员的职责有明确阐述The responsibilities of organiza-tion managers,study director, main researchers, test personnel and quality assurance are clearly described设施设备Facilities and equipment设施环境Facility and environment确保按照标准正常开展Ensure normal operation in ac-cordance with standards仅作原则要求For principle only重视环境条件,尤其是对废弃物处置有明确规定Pay attention to environmental conditions, especially clear regu-lations on waste disposal仪器设备Instruments and equipment强调校准和标准物质包含参考值或修正因子,应确保得到更新和应用It is emphasized that calibration and reference materials contain reference values or correction factors, which should be updated and applied对租用设备开展检验检测活动进行了规定Provisions are made for inspection and testing activities of leased e-quipment更注重按照SOP对仪器进行周期性核查,并且有些仪器还需进行3Q验证Pay more attention to the periodic verification of instruments ac-cording to SOP, and some instru-ments need to be verified by 3Q测量溯源Metrological traceability标准中指定了相关要求Relevant requirements are speci-fied in standard对设备和标准物质的溯源仅作原则要求The traceability of equipment and reference materials is only re-quired in principle强调有关资源溯源信息的可靠性和可用性Emphasize the reliability and a-vailability of relevant resource traceability information试验系统Test system不适用Not applicable不适用Not applicable不相容活动有效隔离,防止交叉污染,更强调生物试验前隔离和条件适应Incompatible activities are effec-tively isolated to prevent cross contamination, with more empha-sis on isolation and condition ad-aptation before biological test
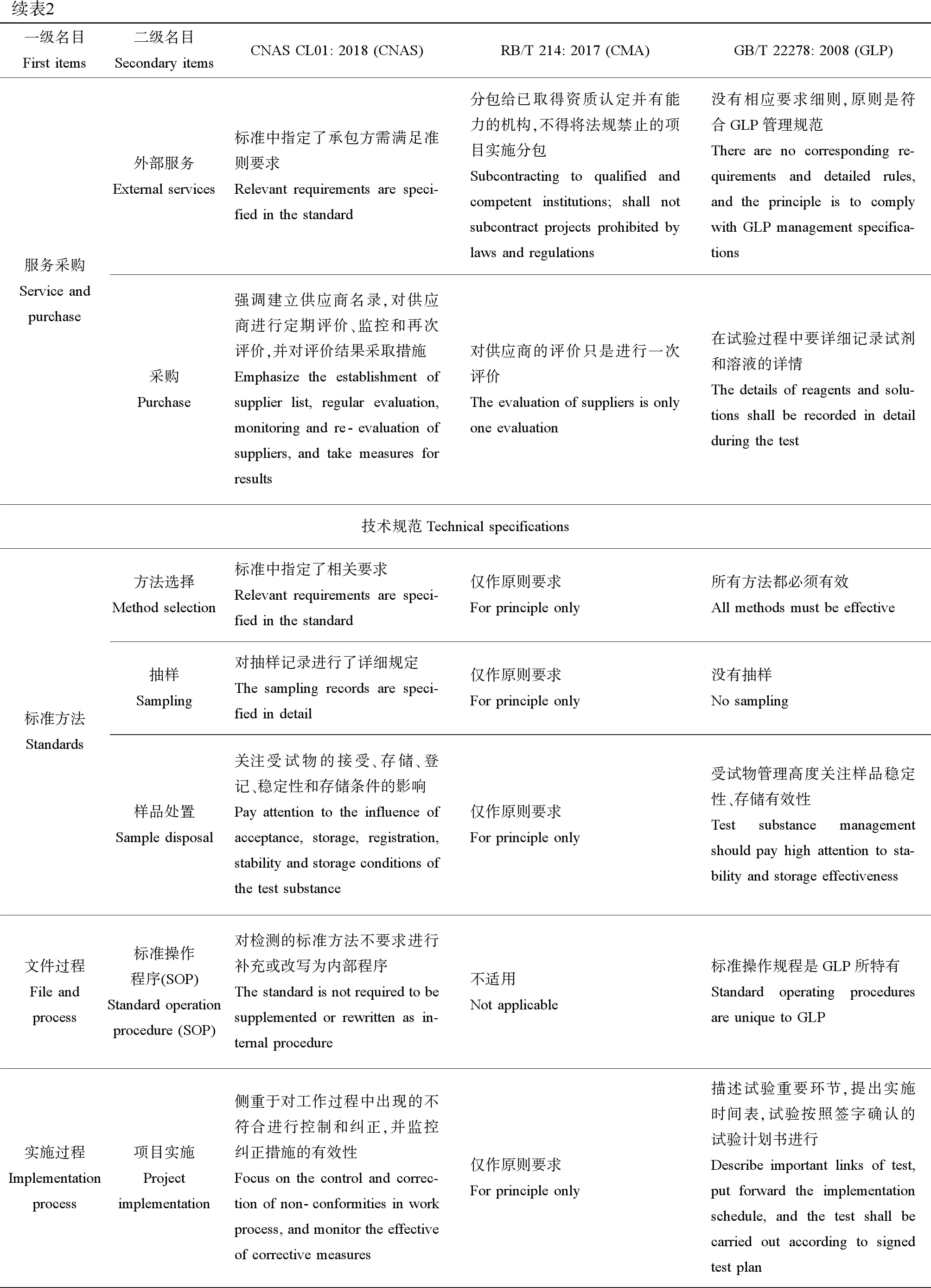
续表2一级名目First items二级名目Secondary itemsCNAS CL01: 2018 (CNAS)RB/T 214: 2017 (CMA)GB/T 22278: 2008 (GLP)服务采购Service and purchase外部服务External services标准中指定了承包方需满足准则要求Relevant requirements are speci-fied in the standard分包给已取得资质认定并有能力的机构,不得将法规禁止的项目实施分包Subcontracting to qualified and competent institutions; shall not subcontract projects prohibited by laws and regulations没有相应要求细则,原则是符合GLP管理规范There are no corresponding re-quirements and detailed rules, and the principle is to comply with GLP management specifica-tions采购Purchase强调建立供应商名录,对供应商进行定期评价、监控和再次评价,并对评价结果采取措施Emphasize the establishment of supplier list, regular evaluation, monitoring and re-evaluation of suppliers, and take measures for results对供应商的评价只是进行一次评价The evaluation of suppliers is only one evaluation在试验过程中要详细记录试剂和溶液的详情The details of reagents and solu-tions shall be recorded in detail during the test技术规范Technical specifications标准方法Standards方法选择Method selection标准中指定了相关要求Relevant requirements are speci-fied in the standard仅作原则要求For principle only所有方法都必须有效All methods must be effective抽样Sampling对抽样记录进行了详细规定The sampling records are speci-fied in detail仅作原则要求For principle only没有抽样No sampling样品处置Sample disposal关注受试物的接受、存储、登记、稳定性和存储条件的影响Pay attention to the influence of acceptance, storage, registration, stability and storage conditions of the test substance仅作原则要求For principle only受试物管理高度关注样品稳定性、存储有效性Test substance management should pay high attention to sta-bility and storage effectiveness文件过程File and process标准操作程序(SOP) Standard operation procedure (SOP)对检测的标准方法不要求进行补充或改写为内部程序The standard is not required to be supplemented or rewritten as in-ternal procedure不适用Not applicable标准操作规程是GLP所特有Standard operating procedures are unique to GLP实施过程Implementation process项目实施Project implementation侧重于对工作过程中出现的不符合进行控制和纠正,并监控纠正措施的有效性Focus on the control and correc-tion of non-conformities in work process, and monitor the effective of corrective measures仅作原则要求 For principle only描述试验重要环节,提出实施时间表,试验按照签字确认的试验计划书进行Describe important links of test, put forward the implementation schedule, and the test shall be carried out according to signed test plan
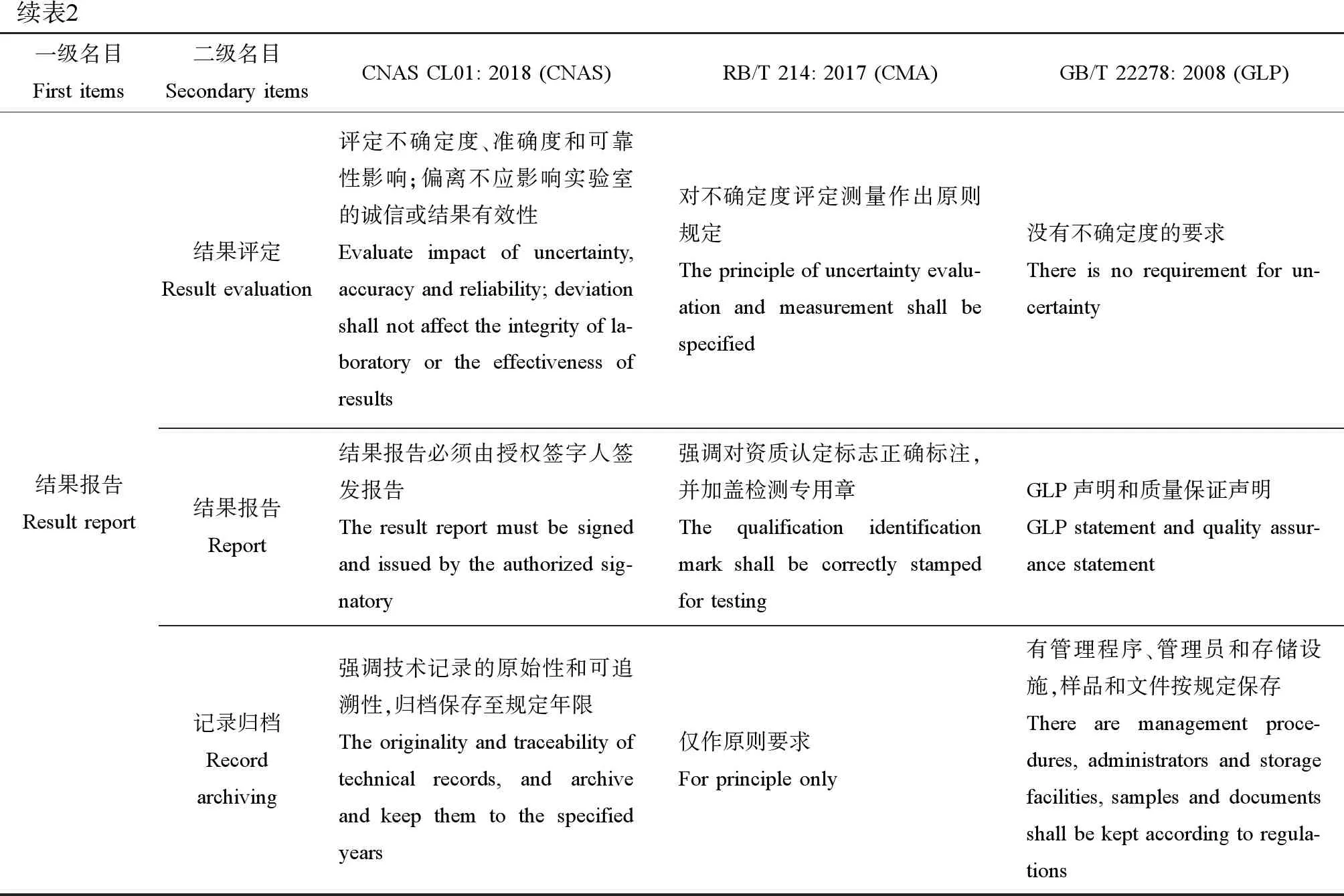
续表2一级名目First items二级名目Secondary itemsCNAS CL01: 2018 (CNAS)RB/T 214: 2017 (CMA)GB/T 22278: 2008 (GLP)结果报告Result report结果评定Result evaluation评定不确定度、准确度和可靠性影响;偏离不应影响实验室的诚信或结果有效性Evaluate impact of uncertainty, accuracy and reliability; deviation shall not affect the integrity of la-boratory or the effectiveness of results对不确定度评定测量作出原则规定The principle of uncertainty evalu-ation and measurement shall be specified没有不确定度的要求There is no requirement for un-certainty结果报告Report结果报告必须由授权签字人签发报告 The result report must be signed and issued by the authorized sig-natory强调对资质认定标志正确标注,并加盖检测专用章The qualification identification mark shall be correctly stamped for testingGLP声明和质量保证声明GLP statement and quality assur-ance statement记录归档Record archiving强调技术记录的原始性和可追溯性,归档保存至规定年限The originality and traceability of technical records, and archive and keep them to the specified years仅作原则要求For principle only有管理程序、管理员和存储设施,样品和文件按规定保存There are management proce-dures, administrators and storage facilities, samples and documents shall be kept according to regula-tions
ISO/IEC 17025与GLP相同之处包括组织机构人员、仪器、受试物参比物、培训、方法有效性和结果报告[29-30]。ISO/IEC 17025和GLP之间除具有部分相同的标准要素,它们还具有各自特有的技术和管理要素。
ISO/IEC 17025实验室的检测项目通常是直接按照客户确认的标准方法开展,不需要制定研究计划和质量保证(quality assurance,QA)现场检查。质量体系独特之处是投诉程序、溯源和不确定度计算、服务客户、预防措施、比对实验和抽样检查。
GLP独特之处是质量保证部门(quality assurance unit,QAU)、QA、标准操作程序(standard operation procedure,SOP)文件、主计划表、研究计划、测试系统、任命项目负责人和档案管理员,仪器强调3Q认证,技术资料与QA资料分开保存等。GLP测试项目必须制定研究计划和SOP,计划实施要求严格按照项目计划书和标准操作规程进行试验操作和现场记录。同时应根据制定的质量保证计划开展包括试验实施、设施设备和研究过程检查。
在实际应用中,一个质量体系不可能代替另外一个,同时运行2种以上质量体系,应综合考虑不同质量体系的区别与联系[31]。
3 关键环节风险识别(Risk identification from key points)
实验室为了维持CNAS、CMA和GLP等各种授权资质,每年都要参加管理部门组织的监督检查,在现场评审中发现的问题,涉及到资源、技术、人员以及信息安全等各个方面。质量体系面临着各个环节的风险考验,不可漫无目的地撒网式识别,实现行之有效的监管和风险全面可控,更多应落实在对关键环节的风险把握[16]。
3.1 17025体系的风险识别
外部评审与内部核查,不仅是17025实验室维系能力授权的方式,也是客观推动质量体系发展的动力[16]。检测实验室每年都要经历各种评审,按照4M1E方法,即从人员(Man)、设备(Machine)、材料(Material)、方法(Method)和环境(Environment)5个方面进行分析(图1),在评审中出现的不符合项涉及组织管理和记录控制条款,还包括人员、检测方法、设备和测量溯源的技术条款。

图1 实验室评审不符合(NCR)影响因素鱼骨图Fig. 1 Fish bone map of non-conforming report (NCR) factors from laboratory review
对不符合项类型的频次分布进行分析(表3)[16],发现17025实验室主要风险点如下:(1)仪器设备的校准验收和测量溯源占设备管理要素58.3%,占总数的37.2%;(2)标准方法、新方法或非标方法的验证记录缺失约占总数12%;(3)人员方面,试验人员能力监督覆盖不全,对标准解读水平差距大,约占总数的9%;(4)管理方面,出现体系性文件规定和标准要求不符约占4%;出现实施性文件规定和实际操作不符约占27%;出现实际操作和目标效果不符约占21%。
综上,管理要素∶技术要素=1∶3,仪器设备∶检测方法∶量值溯源=2∶1∶1。仪器设备管理的规范性出现频次最高,可识别为高风险信号。设备管理的障碍主要有以下几点:(1)仪器设备没有规范的校核程序,无法保证检测结果的可靠性;(2)部分关键设备数量有限,测试任务超负荷运转,不利于提升检测效率;(3)仪器设备备案登记、验收确认、授权操作和分级管理等技术评估工作未能及时完成或认真执行。
3.2 GLP体系的风险识别
根据实验室多年的GLP运行经验,按照管理、技术、仪器设备和检查过程进行总结归纳(表4)[25],在GLP评审中出现的不符合项:技术要素主要涉及试验体系、材料和设备制备和准备、试验操作、试验有效性和试验结果、化学分析和报告完整性5个部分,管理要素主要涉及组织机构人员、质量保证、SOP、样品和档案记录5个部分。

表4 GLP实验室历年定期检查不符合项频次统计表Table 4 Statistics of non-conformities for laboratory GLP inspection over years
在管理方面:没有组织结构图和实验室GLP分区图;关键岗位没有书面任命,或人员资质不合格;人员培训计划和记录过于简单;缺乏系统控制文件,例如计算机和电子文档管理存在安全隐患;管理程序控制不完善,例如测试项目完成后委托方不确认导致项目不能及时归档;管理范围不全面,例如受试物的评估、记录、保存、留样与处置程序不完善导致过期样品无法及时处理。
在技术方面:试验操作的描述过于简单,不能起到指导实际操作的作用;记录内容不全面,例如实际试验操作与SOP和项目计划书有偏差,试验样品前处理的仪器、方法和操作记录不完整;饲料、水和试验生物等方法和检测程序缺乏或不完善,无法对试验材料进行有效管理;现场标识不规范或不完善,导致试验的过程风险;试验过程不全,例如缺少开展难测试物质的设施、方法和验证程序等。
在仪器、设备和设施方面:日常使用、检定、维护和保养等一般都比较完善,但依然时有问题发生。如GLP仪器和非GLP仪器没有进行区分,仪器设备的标识系统不健全,健康和安全防护标识缺乏;大型仪器的数据追踪不完善;仪器没有进行分级管理;定制的测试装置或系统未进行验证;废物处置及仪器设备操作记录不完善。
在SOP和记录档案方面:SOP的批准、分发、修改程序不规范,SOP的不全面、不详细、操作性差,实际操作与SOP不一致;未使用现行有效的SOP和表格;计算机软件使用的功能验证、电子数据的审计追踪等要求缺失或不全面;档案设施不全,档案归档不及时,档案目录不详细,档案借阅不规范等。
在质量保证方面:实施检查依据不完善,没有计划书或修改补充页的副本;交流和追踪记录不完整,发现的问题未及时向项目负责人和机构负责人报告,缺少追踪核查;检查证据留存不完整,质量保证人没有存留项目计划书、测试报告的草稿和终稿,原始记录检查没有存留关键原始记录复印件;分包项目未见QA检查的SOP和检查记录;研究检查中用过程检查代替现场检查。
综上,目前GLP体系的主要风险包括:(1)大型仪器和定制仪器设备需要进行性能验证用于保证仪器的可靠性和稳定性;(2)管理程序和试验操作要有SOP规定,以此保证测试结果的可靠性;(3)记录报告必须满足完整性和准确性的要求,电子数据审计追踪的管理水平需要逐步提高,并达到现代管理发展趋势的要求。
4 风险管理应对措施(Risk management countermeasures)
现代质量保证强调对研究过程进行彻底、深入的检查,而不是通过对单个操作进行快速抽查。检查包括人员、设施设备、标准操作程序、原始数据以及研究过程,研究过程即是完成一个研究阶段所需的所有关键环节,这种方法比在一系列研究中进行肤浅观察更能实现有效的质量保证。
检查的类型取决于研究的性质。在研究期间,每个阶段应该选择影响数据质量的关键环节进行检查。例如,短期研究的关键环节包括准备研究计划书、剂量和试验体系准备、动物分配、试验系统、动物观察、浓度分析、数据记录与统计和最终测试报告。
4.1 风险控制关键环节
我国实验室检测体系发展至今,已进入质量管理全面提升的阶段,规范且可操作的风险管理程序,已成为检测活动的硬性诉求。检测体系中新增的风险控制过程,虽然在不同的实验室活动中形式各异,但大部分实验室通常忽略了关键环节的风险影响。因此,为确保检测实验室风险控制活动的效果,应先从体系关键环节的风险识别入手[16]。
研究发现,实验室风险因素和检测活动所处阶段密不可分,每个阶段主导检测环节主体不同,风险控制内容也不一样。同行评议认可度最高的风险控制3个关键环节,即管理控制(外部评审)、行为控制(报告质量)和技术控制(不确定度评估)[16,32]。外部评审环节是实验室在行业中可信度的体现,报告质量环节直接影响检测的公信力和产品发证,测量不确定度评估环节在专业技术层面影响着前2个环节的控制效果。
实验室质量体系评审的3个风险点分别是[16]:不符合项、报告质量和不确定度评估。报告质量环节主要风险在于原始信息溯源受限,前期检测任务受理阶段和试验阶段记录缺失或矛盾,导致报告环节无法实现可靠的溯源。不确定度的主要风险在于测试系统分量自由度的影响。不符合项和报告质量2个环节所引发的质量问题种类繁多,需要采取硬性管理措施和长期的经验积累进行纠正。
GLP与17025体系虽然在不符合项和报告质量风险控制措施上本质基本相同。然而,GLP实验室不能按照常规方法开展不确定度测量,注重对每一个项目的有效性评估,而17025体系则可以通过不确定度测量评估质量体系的有效性。当然,GLP管理部门可以通过实验室间比对获得不确定度用于技术能力和方法有效性的评估。
4.2 管理可借鉴的方法
同时运维不同质量管理体系,从节约成本和资源角度出发,部分技术要素和管理要素可相互补充,如外部服务和采购、仪器设备、试验系统、人员队伍建设、财务和业务,这几方面没有管理差异,只有要求高低不同,因此可以合并管理,当管理标准不一致时,采用高标准原则能达到统一的质量管理目的。
为确保检测实验室风险控制的效果,质量管理体系建设应先从体系关键环节的风险识别入手,根据质量管理体系差异,区分关键的技术要素和管理要素。
(1)样品管理分开。GLP的样品管理比17025体系要求更加严格,样品除了接收、标记、领用和保存的一般要求外,在使用过程中每次发放都需要核实结余量,样品留样要按照档案管理要求,样品的配制和前处理作为关键环节需要现场核实,样品需在整个产品注册周期内保存,处置时需征得委托方同意。
(2)设施环境分开。GLP测试采用的管理流程和思路与17025体系很不一样,要保证对试验全过程监控,由于人员安排、实验节奏、操作记录和质量控制的要求均不一样,17025与GLP的设施、环境和场所需要分开,不分开势必会相互影响,GLP的完整性有可能受到影响,17025的质量控制有可能会被打断。
(3)记录档案分开。不同质量体系的技术资料和质量资料分开保存和管理,GLP档案设施需要保证安全的存储,研究计划、原始记录、最终报告、样品和试验体系都要求能够溯源,目的是可根据档案资料重建整个检测活动。17025体系的资料并没有如此详尽,它保存相关的原始记录、测试报告、质控证明和样品,只要可以证明检测结果的有效性就可以,这是两者之间的基本差别。
(4)主要人员分开。GLP试验通常都需要2个以上测试人员同时在场进行操作,即需要一人操作一人核对,这样可以保证试验整个阶段都能按照项目计划书执行,测试相关的所有内容都需要SOP和项目计划书及其补充页,任何修改和变动都需要得到认可,技术方面注重项目的执行,测试报告注重试验结果的有效性评价。而17025体系内的人员不需要撰写项目计划书,试验现场没有质量保证人员,他们并不关注过程重建,但注重能力考核和风险整体控制,比如测试标准物质来保证试验结果的可靠性。因此,2种质量管理体系的人员习惯和意识具有很大差异,同时运维2种不同质量管理体系,主要技术人员和质量体系人员一定要分开,这样才能保证测试的顺利进行。
(5)质量管理文件和质量保证职能部门分开。GLP质量体系贯穿整个研究过程,专注于每项研究的执行及过程管理,GLP通过编写和使用标准操作程序规范试验操作和档案记录,通过建立质量保证计划对试验全过程进行QA检查。而17025体系没有标准操作程序的规定,它通过建立质量手册,指定质量主管,并由质量主管按照日程表的要求和管理层的需要组织内部审核来确保检测的可靠性。因此2种体系的质量保证在实施目标、过程和方法上均存在本质差异,只有质量管理文件和质量保证职能部门分开,这样才能保证质量体系相互不干扰。
5 总结(Conclusions)
综上分析,我们发现ISO/IEC 17025与GLP相同之处包括人员仪器、受试物、参比物、培训、方法和报告的基本要求。ISO/IEC 17025特有的要素是投诉程序、溯源和不确定度计算、服务客户、预防措施、比对实验和抽样检查。GLP独特之处是QAU部门、质量保证、SOP文件、主计划表、研究计划、测试系统、项目负责人和档案管理员。
在实际应用中,一个质量体系不可能代替另外一个。同时运行不同的质量体系虽然可以减少资源配置和节约成本,但应综合考虑它们的区别、联系和相容性。人员队伍建设方面,全体技术人员的综合能力考评可以统一组织安排,确保组织技术水平稳步提升。质量保证方面,应明确17025质量体系与GLP质量体系的独立性,GLP体系应能够确保对每一项试验实施有计划的质量监控,17025质量体系则应能满足检测技术能力和公正性的要求。检测活动过程中,不能合并管理的要素包括测试的主要人员、文件体系、测试样品、记录档案、设施和环境。在仪器设备、试验系统、材料试剂和服务采购方面可以资源共享,但合并管理要按照高标准执行。
质量保证部门是负责统筹实验室体系运作的部门,监管工作的效果体现着实验室整体质量风险控制的水平。不同体系相互协调,需要实验室管理部门能够切实理解和应用,正确有效的实施不仅可以推动实验室实现自我提升,在瞬息万变的时代浪潮中,更能为行业的长足发展提供参考。
