炔基改性光固化丙烯酸酯压敏胶
2022-09-26晏虹宇张正源王跃川
晏虹宇, 张正源, 王跃川,2*
1. 四川大学高分子科学与工程学院, 四川 成都 610065;2. 四川大学高分子材料工程国家重点实验室, 四川 成都 610065
辐射固化是以紫外光(UV光)或低能电子束(EB)为能量源的固化技术,与传统、成熟的热固化技术相比,具有固化快、生产效率高、节能、环保等优点,且易与其他技术手段结合实现智能制造。光固化是最易实现、投资小的辐射固化方式,在常规热固化生产线加装紫外灯或UV-LED灯,就可将其改造成光固化生产线,将固化速度提高10余倍,生产线缩短至10 m内。光固化技术已广泛用于涂料、印刷、胶黏剂、光电子器件制造等领域[1]。
压敏胶是一类较轻微施加压力就可产生黏接作用的表面功能材料[2],是胶黏剂的一大类别,广泛应用于标签、贴纸、胶带、医药和电子通信等领域。对不同的应用需求,压敏胶的黏接性能要求差异大,从易黏接、易剥离到永久黏接、耐水、耐候、耐热等。丙烯酸酯压敏胶是用途广、用量大的一类压敏胶,通过长链烷基丙烯酸酯单体与功能性丙烯酸酯单体组合,可以在很宽的范围内设计、调节压敏胶的性能。由于环保和绿色制造的要求,溶液型丙烯酸酯压敏胶面临无溶剂化的升级要求,而乳液型丙烯酸酯压敏胶在性能上仍有不足,需要开发新的环保型压敏胶制备技术[3]。
光固化是实现压敏胶生产无溶剂化的可行途径,也是改进性能的有效手段。光固化压敏胶可通过两种方式来制备,一种方式是热熔UV交联型压敏胶:将溶液聚合制备的树脂脱除溶剂,树脂在高温下涂布,光照交联[4]。为此,丙烯酸酯树脂的分子结构要作调整,以降低树脂的黏度,并在树脂分子骨架上引入光交联功能团。热熔UV交联型压敏胶目前主要用于制备低气味的压敏胶带,已有成功的商品。另一种方式是无溶剂光固化:将丙烯酸酯单体生成高分子量聚合物的过程分解成两步来完成,第一步先用无溶剂光聚合制备低黏度的预聚物——包含高分子量的聚合物和未聚合单体的混合物,第二步是在预聚物中加入功能单体和光引发剂后在涂布线上现场光聚合和光固化。该方法制备的胶液黏度可调,可以涂布到各种基材上,压敏胶厚度易控制,有很大的发展空间。但通常光固化型压敏胶前后两步形成的聚合物分子链主要是物理共混,没有多少化学结合。溶剂型丙烯酸酯压敏胶的性能优异,部分得益于热固化过程中树脂分子链的交联[5],因此,有必要使光固化型丙烯酸酯压敏胶前后两步形成的聚合物分子链间产生一定程度的化学交联。为此,需要在第一步的聚合物分子链上引入可参与光固化的官能团,如果直接用双官能团的丙烯酸酯,预聚合阶段树脂将交联、凝胶化。
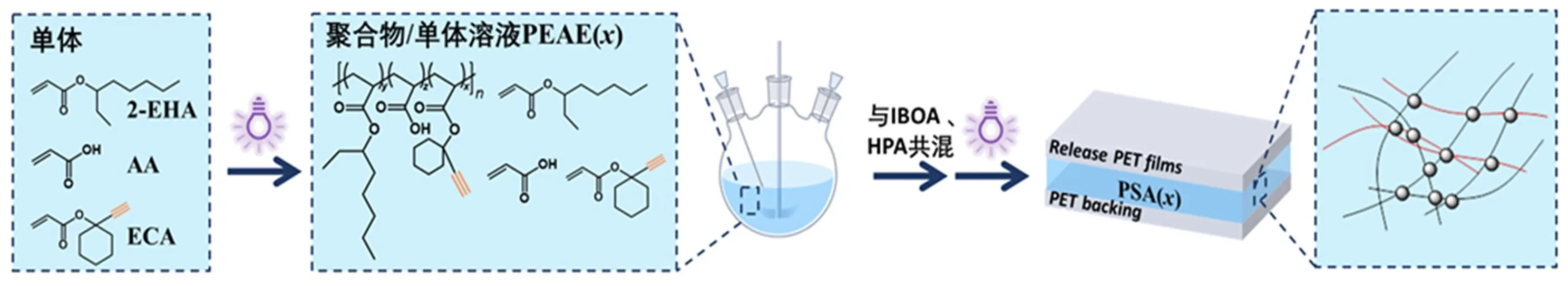
图1 无溶剂光聚合和光固化制备炔基功能化压敏胶
1 实验部分
1.1 实验试剂
二苯基(2,4,6-三甲基苯甲酰基)氧化膦(TPO)、丙烯酸异冰片酯(IBOA)、丙烯酸羟丙酯(HPA)、丙烯酸异辛酯(2-EHA)、丙烯酸(AA),均为市售商品,直接使用。丙烯酸-1-环己基丙炔酯(ECA),自制[7],核磁氢谱(1H NMR)检查纯度>97%。
1.2 炔基功能化丙烯酸酯预聚物PEAE(x)的制备
将单体(2-EHA、AA、ECA)和0.3wt%的TPO加入四口烧瓶中,快速搅拌下通氮气排空气5 min,设定机械搅拌速度250 r/min,用LED灯(365 nm,20 mW/cm2)照射光引发聚合,间隙性光照射以控制反应温度在60 ℃内,反应液逐渐变黏稠,直至轻微爬杆,停止光照,引入空气结束反应,得到可流动、无色透明的炔基功能化丙烯酸酯预聚物PEAE(x),用黑色塑料罐避光常温保存,x为炔基单体的摩尔百分数。
1.3 炔基丙烯酸酯压敏胶PSA(x)的制备
将预聚物PEAE(x)、活性稀释单体IBOA、HPA(质量比74∶20∶6)以及0.3wt%的TPO复配,搅拌均匀并减压消泡,取复配好的胶液倒在50 μm厚PET薄膜上,用刮棒刮涂出50 μm厚的液膜,用离型膜覆盖并置于50 mW/cm2的中压汞灯灯箱中固化,得到以PET膜为背衬的单面可黏接的压敏胶片PSA(x),x为炔基单体的摩尔百分数。测试前,将PSA(x)压敏胶片裁成所需尺寸,撕去离型膜,贴合到待测基材表面并用胶辊辊压,放置30 min后进行测试。
1.4 测试与表征
(1)预聚物中炔基官能团含量:用核磁氢谱(1H NMR)表征预聚物PEAE(x)的化学结构,对峰进行积分计算得到单体的转化率、炔基氢的摩尔百分含量。
(2)分子量及分子量分布:用凝胶色谱法(GPC)测量预聚物PEAE(x)中聚合物的分子量及分子量分布。
(3)压敏胶PSA(x)的玻璃化转变温度:用差示扫描量热仪(DSC)测定压敏胶PSA(x)的玻璃化转变温度,测试程序设定为从25 ℃以40 ℃/min降温至-80 ℃,恒温3 min,再以10 ℃/min升温至80 ℃,获得热流-温度曲线。
(4)180°剥离强度、剪切强度:用万能材料试验机测压敏胶PSA(x)在玻璃基材上的180°剥离强度(拉伸速度300 mm/min)和剪切强度(拉伸速度12.7 mm/min),同一样品平行测3次取平均值。
(5)初黏力:用胶带初黏性试验机(滚球法GB/T 4852—2002)测试压敏胶PSA(x)的初黏力。
(6)持黏力:按标准GB/T 4851—2014制样,以1 kg砝码为载重,测试并记录压敏胶带PSA(x)从玻璃基板上脱落的时间。
2 结果与讨论
2.1 本体光聚合制备预聚物
本工作采用先本体光聚合制备无溶剂预聚物,再加入适量硬单体后进一步光固化的两步光化学方式制备炔基功能化丙烯酸酯压敏胶。第一步光聚合以进行到黏度适宜后光固化为止,得到的预聚物包含高分子量的聚合物和未转化的单体。预聚反应的单体包括主要单体2-EHA与少量AA和炔基功能单体ECA。2-EHA是丙烯酸酯压敏胶的常用单体,Tg低,为第二步光固化加入高Tg的功能单体留下性能调整空间,AA是极性单体,少量加入即可增加压敏胶对极性基材的附着力,ECA作为炔基功能单体,同时调控光聚合反应,见下面的讨论。

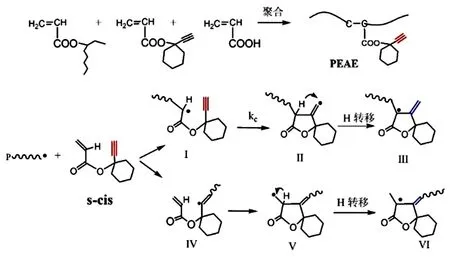
图2 炔基丙烯酸酯单体参与的光聚合反应及环加成和衰减链转移过程
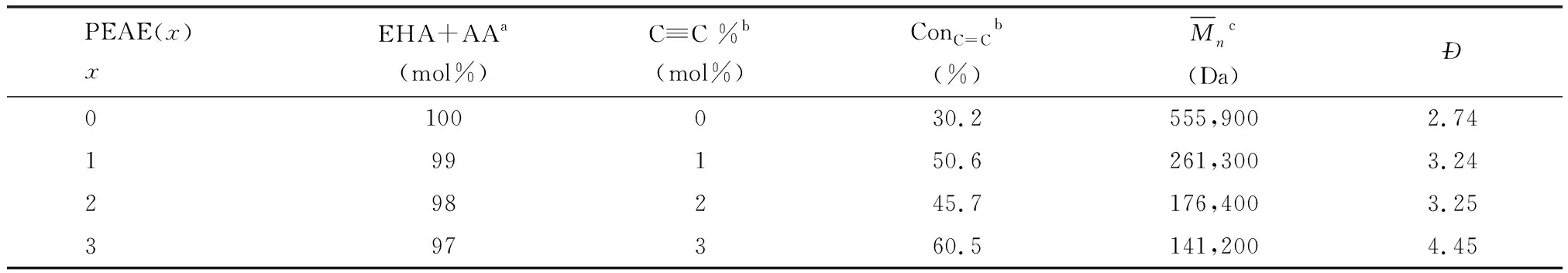
表1 单体投料比及预聚物PEAE(x)的化学结构

图3 不同炔基含量预聚物PEAE(x)的1H NMR谱图和GPC曲线
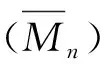
2.2 炔基对压敏胶PSA(x)性能的影响
在预聚物PEAE(x)中加入功能单体IBOA、HPA和光引发剂TPO配制成可光固化的压敏胶胶液,在光辐射作用下,TPO引发单体进行自由基聚合反应,直至固化。在此聚合、固化过程中,PEAE(x)分子链上的部分C≡C以及少量环化加成反应后的不饱和内酯也会与自由基反应,使光固化阶段的聚合物分子链与预聚物中的聚合物分子链交联。对PEAE(0),此时的光固化主要是单体聚合生成高聚物的过程。比较PSA(0)与PSA(x)(x=1, 2, 3)的性能差异,可以推断炔基官能团对光固化交联以及压敏胶性能的影响。
压敏胶的黏性和抗剥离性能,是柔软、易流动的聚合物分子链段、高分子链间多而弱的相互作用,以及量少但较强的极性基团间相互作用共同作用的结果。性能优异的压敏胶是综合了树脂性能、单体结构后精细配制的产物,本工作主要是考察炔基功能单体对压敏胶的影响,无意优化压敏胶的配方和性能,上述单体与树脂的组成仅是一个用于对比的基本配方。
图4(a)是光固化压敏胶PSA(x)的DSC测试曲线,由于ECA均聚物的Tg较高,约110 ℃[6],在ECA共聚物中起到硬单体的作用,在实验研究的范围内,PSA(x)的Tg随x的增加而略有增加,ECA含量最高的PSA(3),其Tg低于-18 ℃,与大多数丙烯酸酯压敏胶的Tg类似。
光固化时间(30 s、90 s、180 s)对PSA(x)的180°剥离力的影响如图4(b)所示。PSA(0)和PSA(1)由于交联度不够,即使固化180 s,剥离时仍发生内聚破坏,剥离强度为625~869 N/m,与典型的丙烯酸酯压敏胶的性能大致相当。PSA(2)和PSA(3)炔基含量更高,在曝光能量不足时(光照30 s),由于固化反应不完全,剥离面呈内聚破坏,且剥离力与同样光照能量后的PSA(0)、PSA(1)相比较小,这可能是炔基含量增加,聚合反应过程中衰减链转移增加,预聚物分子量明显降低导致的。增加光固化能量,即光照时间延长后(光照90 s、180 s),PSA(2)和PSA(3)的剥离强度都明显上升,达到774~809 N/m,与PSA(0)和PSA(1)的大致相当,但剥离面在玻璃界面,表明炔基参与光反应,导致了分子链间交联,提高了胶层的内聚强度。
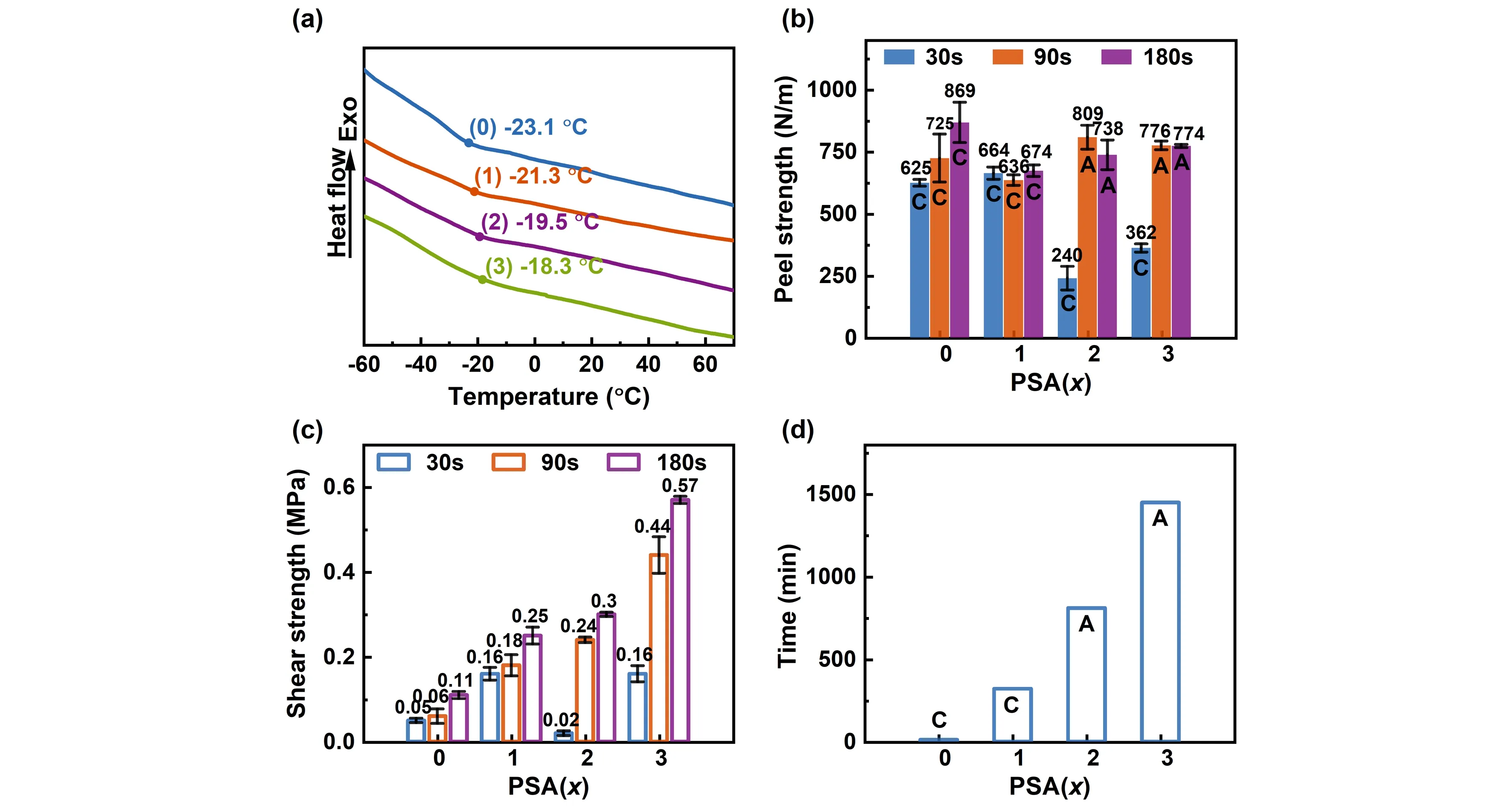
图4 光固化压敏胶PSA(x)的DSC曲线(a)、180°剥离力(b),动态(c)和静态(d)剪切性能破坏类型:A为界面破坏、C为内聚破坏
采用滚球法评价了180 s光固化的PSA(x)的初黏力,不同炔基含量的PSA(x)的初黏力均处在同一水平,均能黏住8#不锈钢小球,其中PSA(2)和PSA(3)的初黏力略高,说明虽然ECA含量增加导致Tg增大,但并未对PSA(x)的初黏力产生负面影响,反而有所增加。
考察了不同光固化时间(30 s、90 s、180 s)制备的压敏胶PSA(x)贴合玻璃基板的动态抗剪切强度差异。如图4(c),剪切强度随着光固化时间的增加均上升。在光固化时间不足时(30 s),由于固化反应不完全,PSA(x)的剪切强度与x含量相关性不强,延长光照时间至90 s和180 s,PSA(x)的固化程度提高,炔基能更多地参与交联,剪切强度明显提高,炔基含量x与PSA(x)剪切强度呈正相关。光固化180 s得到的PSA(3),剪切强度达到0.57 MPa,为PSA(0)的5倍。
用悬挂固定载荷的方法比较了光固化180 s制备的PSA(x)在室温下(25 ℃)下抗静态剪切的性能,如图4(d)示,PSA(0)由于未交联,仅10 min砝码即滑脱,发生内聚破坏;同样的原因,PSA(1)由于交联度较低,在300 min时发生内聚破坏,在PET基膜以及玻璃基板上均有残胶;炔基含量更高的PSA(2)和PSA(3)分别在800 min、1450 min发生界面破坏,玻璃基板上无残胶,相对PSA(0),保持力增加了80倍和145倍。PSA(x)抵抗静态剪切的性能明显上升,压敏胶从内聚破坏转为界面破坏,说明增加炔基含量提高了PSA(x)的内聚强度。该结果与PSA(x)动态剪切强度以及180°剥离力的结果一致。
3 结语
压敏胶的性能是树脂分子的化学组成、分子量、分子链间的物理和化学交联、分子链的柔软性、自由体积等因素共同导致的结果,过强的分子链间相互作用会影响分子链段运动,会制约压敏胶黏力、压敏性。本工作结果表明,利用炔基功能化丙烯酸酯的调控作用,通过无溶剂光聚合可得到流动性好、黏度适宜的炔基功能化丙烯酸酯预聚物,进一步光固化可以制得炔基功能化丙烯酸酯压敏胶;利用炔基参与的光固化可以改善光固化压敏胶的内聚强度,而对其他性能影响不大。由此,丙烯酸酯压敏胶多了一个新的改性手段。此外,还可以通过热或光催化的巯-炔“点击”化学对光交联的炔基功能化压敏胶进行表面改性,这可能是开发特种光固化材料的技术切入点。
