十种食源性致病菌的多重PCR快速检测方法
2022-09-16张明娟刘明明余秋地袁磊王娟秦爱李根容
张明娟,刘明明,余秋地,袁磊,王娟,秦爱,李根容
(重庆市计量质量检测研究院,重庆,400020)
食源性疾病是指通过摄食而进入人体的有毒有害物质等致病因子所造成的疾病,是食品安全的主要问题之一,世界卫生组织数据显示,全球平均每年有上亿腹泻病例,食源性疾病患者数以亿计,其中超70%食源性疾病由致病微生物引起[1-2]。因此,食源性致病菌仍是目前引起食源性疾病的主要原因,如何准确、快速、灵敏地检测食源性致病菌已成为控制食品安全问题的关键[3]。
目前,我国开展食品中食源性致病菌监测工作主要涉及的检测指标有沙门氏菌属、金黄色葡萄球菌、志贺氏菌属、单核细胞增生李斯特氏菌、副溶血性弧菌、大肠埃希氏菌O157∶H7、克罗诺杆菌属、铜绿假单胞菌、粪链球菌、产气荚膜梭菌等,依据GB 4789系列和GB 8538标准,检测方法主要是传统培养法,包括细菌培养、生理生化鉴定等过程,其检测周期较长,操作较繁琐复杂,且检测时易受微生物生长情况影响,上述方法已不能满足我国公共卫生突发事件应急检测的需要[2,4],因此,建立一种高通量、准确、快速的致病菌检测方法显得十分重要。
多重PCR是在常规PCR基础上发展起来的,其反应原理、反应试剂和操作过程与常规PCR相同,区别在于多重PCR技术在同一个反应体系中加入两对或以上引物,分别扩增不同的模板,得到不同的目的片段,实现了一次性检测多个基因的目的[5-7],可以节省检测时间和费用成本,因此具有很好的发展前景。本研究旨在建立一种高通量、快速、准确、灵敏的十重PCR检测方法,在同一反应体系中同时检出沙门氏菌属、金黄色葡萄球菌、志贺氏菌属、单核细胞增生李斯特氏菌、副溶血性弧菌、大肠埃希氏菌O157:H7、克罗诺杆菌属、铜绿假单胞菌、粪链球菌和产气荚膜梭菌,以应用于实际检测工作,解决现有技术操作较为繁琐且耗时长的问题,为监管部门开展食源性致病菌监测提供技术支持。
1 材料与方法
1.1 材料与试剂
1.1.1 实验样品与菌株
预包装的牛肉干、海苔、巧克力、面包、椰汁、鸡精、奶粉、矿泉水样品,市售。实验用菌株见表1。
1.1.2 试剂
10×PCR buffer、2×Multiplex PCR Master Mix、d NTPs、TaqDNA聚合酶、HSTaqDNA酶、Mg2+(浓度25 mmol/L)、DNA Marker(100、50 bp)、ddH2O、琼脂糖、50×TAE、核酸染料、溶菌酶、6×甘油凝胶上样缓冲液,生工生物工程(上海)股份有限公司;细菌基因组提取试剂盒,北京天根生化科技有限公司;营养琼脂、胰酪胨大豆酵母浸膏琼脂、脑心浸液肉汤、细菌琼脂粉、胰蛋白胨大豆琼脂、无菌脱纤维羊血、蛋白胨、缓冲蛋白胨水、7.5% NaCl肉汤、LB肉汤、改良EC肉汤、3% NaCl碱性蛋白胨水、营养肉汤、志贺氏菌增菌液、肠道增菌液、平板计数琼脂,北京陆桥技术股份有限公司;牛肉浸粉,青岛海博生物技术有限公司;NaCl,重庆川东化工(集团)有限公司。

表1 实验菌株名称、编号及来源Table 1 Name, number and source of experimental strains
1.2 仪器与设备
TCEN-24R台式高速冷冻离心机、MiniT-H3金属浴,杭州奥盛仪器有限公司;BJC2-60L-D超纯水仪,重庆华创水处理工程有限公司;Gel DocTMEZ凝胶成像系统、DowerDacTMBasic电泳仪及水平电泳槽、T100型PCR仪,美国Bio-Rad公司;NanoDrop ONEc超微量分光光度计,美国Thermo公司;DW-86L388A超低温冰箱,北京海尔公司;DRP-9272电热恒温培养箱,上海森信实验仪器有限公司;MIX-3000旋涡混合器,上海Gallop Instruments公司;ME403电子天平,瑞士Mettler-Toledo公司;AC2-6S1生物安全柜,新加坡ESCO公司;SQ810C高压灭菌锅,重庆雅马拓科技有限公司;7 L MGC厌氧培养盒,日本三菱公司。
1.3 实验方法
1.3.1 基因组DNA提取
采用细菌基因组提取试剂盒提取细菌基因组DNA。
1.3.2 引物设计及合成
分析细菌种属间的差异,选择特异的保守基因做为检测目的基因[8-21];在美国国立生物技术信息中心(National Center for Biotechnology Information, NCBI)中GenBank数据库上下载目的基因的核酸序列,将目标基因的核酸序列与NCBI数据库进行BLAST比对,验证其特异性;通过引物设计软件Premier 6.0针对其目的基因的序列差异处设计引物;选择特异性较高、长度在18~30 bp、退火温度Tm值接近、引物间二级结构较少且十对引物所扩增的产物长度差异不低于50 bp的引物组,引物组信息见表2。引物由生工生物工程(上海)股份有限公司代为合成,引物纯化方式为iPAGE,引物合成之后用ddH2O稀释到浓度10 μmol/L,-20 ℃保存备用。

表2 十对引物组、目的基因和扩增产物长度Table 2 Ten pairs of primer sets, target genes and amplification product length
1.3.3 引物特异性验证
以表1中标准菌株的基因组DNA为模板,分别对上述10对引物进行单重PCR特异性验证。PCR反应体系:10×PCR buffer 2.5 μL,正反引物(10 μmol/L)各0.5 μL,dNTPs(10 mmol/L)0.5 μL,Taq酶(5 U/μL)0.5 μL,DNA模板1.0 μL,Mg2+(25 mmol/L)1.5 μL,ddH2O补充至50 μL。PCR反应条件:95 ℃预变性5 min;95 ℃变性30 s,57 ℃复性30 s,72 ℃延伸1 min,循环35次;72 ℃延伸10 min。PCR扩增产物取8 μL用于琼脂糖凝胶电泳分析(质量分数为1.5%),并将PCR产物进行测序以验证扩增的准确性。
1.3.4 引物灵敏性验证
以标准菌株铜绿假单胞菌ATCC 27853、单核细胞增生李斯特氏菌ATCC 19115、粪链球菌ATCC 29212、产气荚膜梭菌ATCC 13124、肠炎沙门氏菌CICC 21482、阪崎克罗诺杆菌CICC 21544、福氏志贺氏菌CICC 21534、金黄色葡萄球菌CICC 21600、副溶血性弧菌CICC 21617、大肠埃希氏菌O157∶H7 CICC 21530的基因组DNA为模板,用ddH2O进行10倍梯度稀释,并进行相应的单重PCR体系扩增,验证引物的灵敏度。
1.3.5 多重PCR条件优化
以标准菌株铜绿假单胞菌ATCC 27853、单核细胞增生李斯特氏菌ATCC 19115、粪链球菌ATCC 29212、产气荚膜梭菌ATCC 13124、肠炎沙门氏菌CICC 21482、阪崎克罗诺杆菌CICC 21544、福氏志贺氏菌CICC 21534、金黄色葡萄球菌CICC 21600、副溶血性弧菌CICC 21617、大肠埃希氏菌O157∶H7 CICC 21530的基因组DNA为模板构建多重PCR反应体系。多重PCR反应基本体系:2×Multiplex PCR Master Mix 25 μL,正反引物(10 μmol/L)各0.5 μL,HSTaqDNA酶(5 U/μL)1 μL,DNA模板均1.0 μL,补充ddH2O至50 μL;多重PCR反应基本条件:95 ℃预变性5 min;95 ℃变性30 s,58 ℃退火30 s,72 ℃延伸1 min,循环35次;72 ℃延伸10 min。琼脂糖凝胶电泳分析条件:核酸染料10 μL/100 mL,电泳液1×TAE,PCR扩增产物上样量25 μL,电压220 V,电泳时间约40 min。
1.3.5.1 退火温度优化
在多重PCR反应体系中同时加入10对引物组和10种标准菌株基因组模板DNA,分别设置退火温度为55.0、55.7、56.9、58.8、61.1、63.0、64.3、65.0 ℃进行多重PCR反应。
在多重PCR反应体系中同时加入10对引物组,分别加入1种标准菌株基因组模板DNA,进行多重PCR反应。
1.3.5.2 引物浓度优化
采用优化的退火温度及固定的模板量(50 ng),分别设置引物浓度为20、40、60、80、100 nmol/L进行多重PCR反应。
1.3.5.3 模板量优化
采用优化的退火温度及引物浓度,分别设置模板量为10、25、50 ng进行多重PCR反应。
1.3.6 多重PCR体系特异性验证
以标准菌株的基因组DNA为模板,依据现行标准中各类样品需检测的致病菌类型组合,分组加入10对特异性引物进行以优化后的多重PCR反应体系及条件多重PCR扩增,分组信息见如表3。

表3 致病菌类型组合分组Table 3 Grouping of pathogen type combinations
1.3.7 多重PCR体系灵敏度验证
分别以标准菌株铜绿假单胞菌ATCC 27853、单核细胞增生李斯特氏菌ATCC 19115、粪链球菌ATCC 29212、产气荚膜梭菌ATCC 13124、肠炎沙门氏菌CICC 21482、阪崎克罗诺杆菌CICC 21544、福氏志贺氏菌CICC 21534、金黄色葡萄球菌CICC 21600、副溶血性弧菌CICC 21617、大肠埃希氏菌O157∶H7 CICC 21530的基因组DNA为模板,用ddH2O进行10倍梯度稀释,以优化后的多重PCR反应体系及条件进行多重PCR,验证体系的灵敏度。
1.3.8 多重PCR检测方法初步应用
不同基质对于细菌的培养以及基因组DNA提取均会产生不同影响,选取不同基质、不同污染菌株的人工污染的样品进行实验,进一步验证该多重PCR方法检测的灵敏度。
1.3.8.1 人工污染菌株培养及计数
将标准菌株铜绿假单胞菌ATCC 27853、单核细胞增生李斯特氏菌ATCC 19115、粪链球菌ATCC 29212、产气荚膜梭菌ATCC 13124、肠炎沙门氏菌CICC 21482、阪崎克罗诺杆菌CICC 21544、福氏志贺氏菌CICC 21534、金黄色葡萄球菌CICC 21600、副溶血性弧菌CICC 21617、大肠埃希氏菌O157:H7 CICC 21530分别培养24 h,挑取菌落于无菌水中制成菌悬液,在相应的培养条件下培养48 h,对菌悬液进行平板计数。
1.3.8.2 人工污染方法
用无菌水分别将10株菌稀释至106、105、104、103CFU/mL 4个浓度,每个稀释度分别加入1 mL到10 g基质中,补充增菌液体积至90 mL,具体分组及培养信息见表4。增菌后(36 ℃,24 h)分别吸取1 mL提取基因组DNA,以优化后的多重PCR反应体系及条件进行多重PCR,验证灵敏度。
表4 人工污染实验分组Table 4 Grouping of industrial pollution experiments
2 结果与分析
2.1 引物特异性
通过设计的10对特异性引物对表1中菌株进行PCR检测,结果表明1株单核细胞增生李斯特氏菌菌株、7株沙门氏菌属菌株、7株克罗诺杆菌属菌株、4株志贺氏菌属菌株、1株金黄色葡萄球菌菌株、1株副溶血性弧菌菌株、1株大肠埃希氏菌O157∶H7菌株、1株铜绿假单胞菌菌株、1株粪链球菌菌株、5株产气荚膜梭菌种均扩增出目的条带,而其他阴性对照菌株均未扩增出明显的目的条带,详见电子增强出版附件(http://doi.org/10.13995/j.cnki.11-1802/ts.031352)。
PCR产物进行凝胶电泳验证后,将其送到生工生物工程(上海)股份有限公司代为测序,测序结果通过与目的基因进行序列对比,相似率均达到98%以上,扩增产物均与目标基因序列高度一致。结果表明本研究所设计的10对引物具有高度特异性。
2.2 引物灵敏度
通过PCR检测,单核细胞增生李斯特氏菌、金黄色葡萄球菌、志贺氏菌属、副溶血性弧菌、铜绿假单胞菌、粪链球菌、产气荚膜梭菌、沙门氏菌属、克罗诺杆菌属、大肠埃希氏菌O157∶H7菌株的灵敏度分别为5.37×10-6、5.37×10-4、6.04×10-6、8.85×10-3、9.52×10-6、8.91×10-7、7.33×10-6、7.10×10-3、7.49×10-7、7.20×10-3ng/μL,引物均具有较高灵敏度。
2.3 多重PCR条件优化
2.3.1 退火温度优化
当退火温度设置为55~65 ℃时,多重PCR均能扩增出10条特异性目标条带,见图1-a。
选择55~65 ℃的平均值60 ℃作为退火温度,在多重PCR反应体系中同时加入10对引物组,分别加入1种标准菌株基因组DNA,进行多重PCR反应,在仅加入粪链球菌、沙门氏菌、志贺氏菌基因组DNA时出现非特异扩增,结果见图1-b,因此,60 ℃作为退火温度时,多重PCR体系特异性较差;将退火温度增加至62 ℃,在多重PCR反应体系中同时加入10对引物组,分别加入1种标准菌株基因组DNA,进行多重PCR反应,结果见图1-c,10对特异性引物均只针对加入的模板扩增出特异性条带,无非特异扩增,多重PCR体系特异性较好。因此,综合选择62 ℃作为本研究多重PCR体系的退火温度。

a-温度梯度PCR结果(M-100 bp Marker;1~8分别为55.0、55.7、 56.9、58.8、61.1、63.0、64.3、65.0 ℃);b-退火温度60 ℃; c-退火温度62 ℃(M-100 bp Marker;1~11依次为大肠埃希氏菌 O157∶H7、粪链球菌、克罗诺杆菌属、铜绿假单胞菌、 副溶血性弧菌、产气荚膜梭菌、金黄色葡萄球菌、单核细胞增生 李斯特氏菌、沙门氏菌属、志贺氏菌属、CK)图1 退火温度优化结果Fig.1 Results of annealing temperature optimization
2.3.2 引物浓度优化
在退火温度为62 ℃和模板量为50 ng时,不同引物浓度进行多重PCR检测,结果见图2,当引物浓度为20 nmol/L时,无法完全扩增出10个目的条带,引物浓度为40 nmol/L时,可扩增出10个条带,但条带较模糊,引物浓度在60~100 nmol/L时,均可扩增出10条特异性目标条带,引物浓度在100 nmol/L时,10个目标条带均最清晰,因此,综合选择100 nmol/L作为本研究多重PCR体系的最佳引物浓度。

a-引物浓度20 nmol/L;b-引物浓度40 nmol/L;c-引物浓度60 nmol/L;d-引物浓度80 nmol/L;e-引物浓度100 nmol/L图2 不同引物终浓度的多重PCR扩增结果Fig.2 Multiplex PCR amplification results of different primer final concentrations注:M-100 bp Marker;泳道1、2为2个平行;泳道3为空白(下同)
2.3.3 模板量优化
在退火温度为62 ℃引物浓度为100 nmol/L时,用不同模板量进行多重PCR检测,当模板量加入10 ng时,可扩增出10个条带,但分子量较低的条带较模糊,当模板量为25、50 ng均可扩增出清晰的产物条带,模板量为50 ng时,10个目标条带均最清晰,结果见图3。因此,综合选择50 ng作为本研究多重PCR体系的最佳模板量。
2.4 多重PCR体系特异性验证
1~8组分组实验结果中,均只扩增了目标菌特异性条带,无非特异扩增,结果见图4。因此,本研究建立的多重PCR检测体系具有较好的特异性。

a-模板量10 ng;b-模板量25 ng;c-模板量50 ng图3 不同模板量的多重PCR扩增结果Fig.3 Multiplex PCR amplification results with different template amounts

a~h分别为致病菌分组1~8图4 多重PCR体系特异性验证结果Fig.4 Verification results of specificity of multiplex PCR systems注:M-100 bp Marker;泳道1~3为3个平行
2.5 多重PCR体系灵敏度验证
单核细胞增生李斯特氏菌、金黄色葡萄球菌、副溶血性弧菌、粪链球菌、产气荚膜梭菌、沙门氏菌属、克罗诺杆菌属、志贺氏菌属、大肠埃希氏菌O157∶H7、铜绿假单胞菌菌株的灵敏度分别为5.37×10-2、5.37×10-2、8.85×10-1、8.91×10-1、7.33×10-1、7.10×10-1、7.49×10-2、6.04×10-2、7.20×10-2、9.52×10-2ng/μL,所有菌株的灵敏度均达到10-1ng/μL,该多重PCR体系具有较高的灵敏度。
2.6 多重PCR检测方法初步应用
通过人工污染样品评价实验发现,本研究涉及的10种致病菌在增菌后检测灵敏度均能达到10 CFU/mL以上,且扩增条带清晰明显,扩增效果好,结果见图5。本研究建立的十重PCR检测方法对食品样品进行前增菌处理后,可满足检验需要。
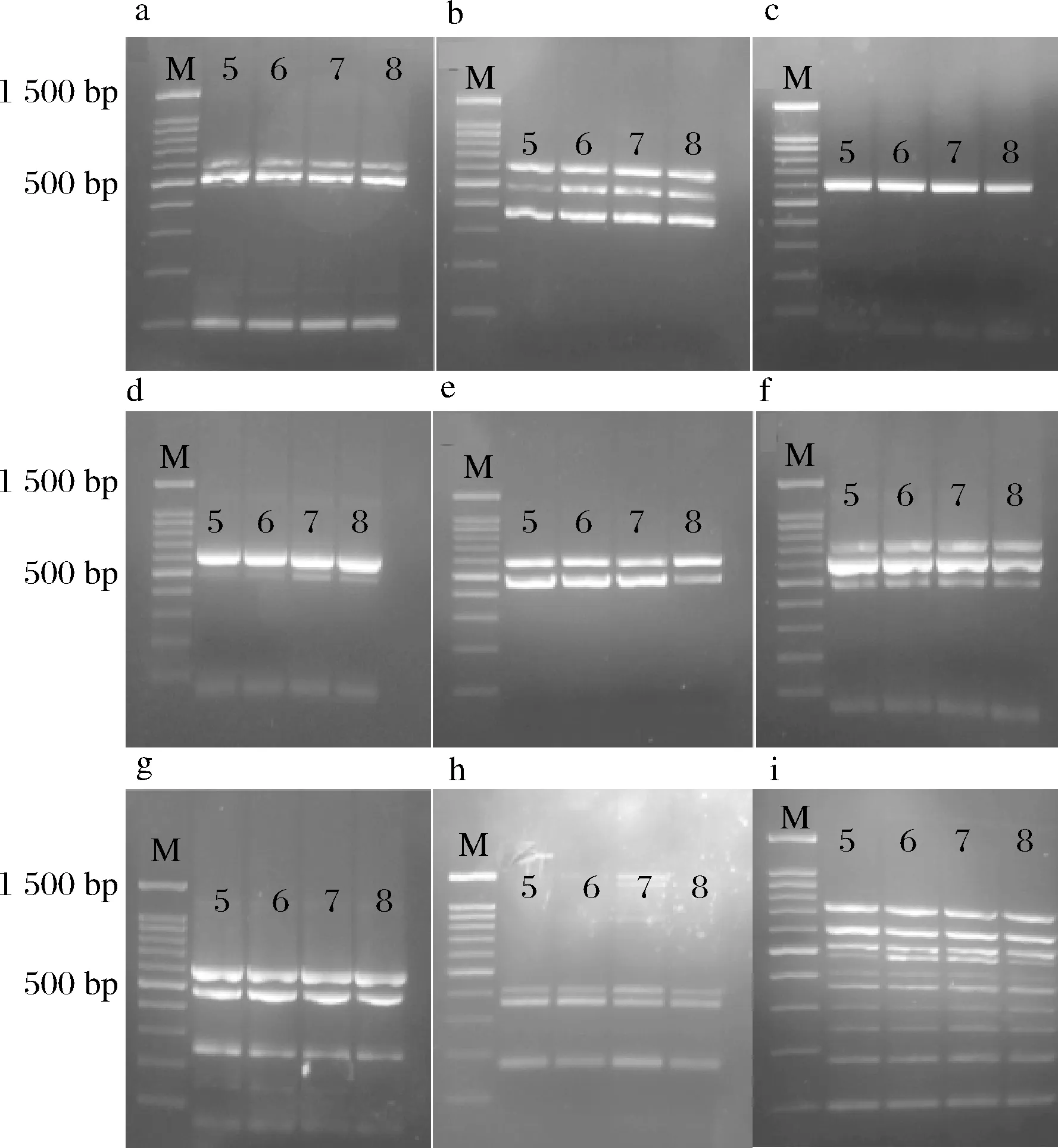
a~i分别为人工污染样品组1~9图5 人工污染样品灵敏度评价结果Fig.5 Evaluation results of sensitivity of artificially contaminated samples注:M-100 bp Marker;泳道5~8-菌液浓度为101、 102、103、104 CFU/mL
3 结论与讨论
本研究建立了一种能够在同一PCR反应体系中同时检测单增细胞增生李斯特菌、沙门氏菌属、克罗诺杆菌属、志贺氏菌属、金黄色葡萄球菌、副溶血性弧菌、大肠埃希氏菌O157∶H7、铜绿假单胞菌、粪链球菌、产气荚膜梭菌的多重PCR检测方法,该方法具有快速、准确、实用性强的优点。优化后多重PCR检测体系的退火温度为62 ℃,最佳模板量为50 ng,最佳引物浓度为100 nmol/L,在此优化条件下,多重PCR反应体系对10种致病菌的检出限均可达10-1ng/μL,此外,应用于人工污染样品检测,该方法经过简单增菌后,10种致病菌的检出限均可达10 CFU/mL,而一般食品受微生物污染后,大多会呈对数生长,繁殖较快,受污染的食品中微生物量极易达到或超过10 CFU/mL,且多数致病菌需要达到一定的数量才能致病,如GB 29921—2021《食品安全国家标准 预包装食品中致病菌限量》中金黄色葡萄球菌的最大可允许超出值多为100 CFU/g,因此,该方法可满足目前食品中食源性致病菌快速检测需要。
相比于普通PCR方法,本研究建立的多重PCR检测方法可以一次反应检测10种致病菌,极大地提高了检测效率,降低了大量的人力、时间、试剂成本。相比于荧光定量PCR方法,本研究建立的方法所需仪器及试剂费用上具有较大优势,不需要合成价格较贵的探针,检验成本可减少2倍左右,且荧光定量PCR方法由于受荧光检测通道的影响,一般只能同时扩增至四重或者五重,使其在致病菌筛选中的应用受到局限,无法满足高通量检测的需求,本研究建立的多重PCR检测方法可避免此局限性。
本研究建立的多重PCR检测方法只需要1次人工操作和1次PCR反应,可将传统微生物检测中所需时间从2~7 d缩短至48 h内,对于应对食品安全突发事件,具有十分突出的意义,且本方法可同时快速检测目前食品安全抽检工作中涉及的10种食源性致病菌,与实际检测工作联系十分紧密,实用性较高,仪器、试剂成本较低,操作较简便,本方法在国内实验室较易普及推广,具有较高的应用价值。
本研究建立的多重PCR检测方法仍需要通过增菌后才能实现检测,这一步严重影响了多重PCR检测效率,因此后期研究中需结合目标菌的富集技术,以进一步提高检测效率,如磁珠富集和双抗体富集技术等。此外,随着多重PCR体系中引物对的增加,PCR产物条带的不断增加,为后续产物分离带来了难点,因此多重PCR技术也可以结合除电泳技术以外的产物分离检测技术,如发展比较成熟的液相、液质技术等,以扩大多重PCR检测方法的应用范围。
