染色质免疫共沉淀测序技术研究进展
2022-09-14陈桂芳杨佳怡高运华任歌
陈桂芳 杨佳怡 高运华 任歌
(1.中国计量科学研究院,北京 100029;2.沈阳化工大学,沈阳 110142)
组蛋白修饰和转录因子对基因表达具有重要的调控作用。在真核细胞中,DNA缠绕着由H2A、H2B、H3、H4构成的核心组蛋白八聚体形成核小体,通过连接DNA串联形成染色质。组蛋白修饰的发生将影响组蛋白与DNA的亲和性,改变染色质的可及性,进而影响基因的表达。转录因子识别并结合基因上游区域特定DNA序列,调控相关基因的表达。染色质免疫共沉淀(chromatin immunoprecipitation,ChIP)利用目的蛋白特异性抗体,与可溶性染色质免疫共沉淀,特异性地富集目的蛋白结合的DNA。与芯片技术相结合的ChIP-chip(chromatin immunoprecipitation-chip)是利用表面覆盖已知序列的核苷酸探针的芯片,对染色质免疫共沉淀捕获的DNA进行核酸杂交,通过传感器检测碱基互补配对产生的荧光信号,进一步分析目的蛋白结合位点[1]。ChIP-chip检测覆盖率受限于芯片上预先设定的DNA序列,存在分辨率低、灵敏度有限,对探针设计要求高等局限性[2]。随着高通量测序技术的发展以及测序成本不断降低,染色质免疫共沉淀与测序相结合的ChIP-seq被广泛使用。研究人员利用生物信息学工具对高通量测序生成的大量数据进行分析,通过将测序序列比对到全基因组,定位转录因子结合、组蛋白修饰的区域[3]。近年来,ChIP-seq被越来越广泛地用于拟南芥、水稻及其他植物的基因表达调控研究中。
ChIP-seq通常需要数百万个细胞,染色质免疫共沉淀和测序文库制备包含多个实验步骤,免疫共沉淀可能受到非特异性结合的影响,产生背景噪音,文库中DNA中GC含量过高或过低,将导致PCR扩增偏倚,进而影响分析结果的准确性。研究人员利用流式细胞术、微流控芯片等技术分离少量细胞或单细胞,优化染色质片段化、免疫共沉淀以及测序文库构建等实验流程;通过特异性抗体引导将MNase或Tn5转座酶间接结合到目的蛋白,并在蛋白结合位点附近使染色质断裂,替代了ChIP-seq染色质片段化和免疫共沉淀操作,简化实验操作流程。在上述基础上,实现少量细胞或单细胞水平的ChIP-seq检测。本文简述了ChIP-seq原理,详细介绍其数据分析方法,讨论近年来发展的ChIP-seq优化方法和衍生技术,分析并比较不同方法的特点,总结了植物转录因子和组蛋白修饰在生物钟调控、激素信号转导、光信号途径、胁迫响应等方面研究与应用。
1 ChIP-seq原理及数据分析
1.1 ChIP-seq原理
组蛋白修饰主要发生在组蛋白的N端,核小体组蛋白被DNA环绕,两者结合较稳定。转录因子一般具有DNA结合结构域,识别靶基因并以序列特异性方式结合DNA,转录因子与DNA相互作用通常是动态的[4-5]。根据目的蛋白与DNA结合特性,ChIP中制备染色质片段的方式不同,主要包括甲醛交联染色质免疫共沉淀(formaldehyde cross-linking and sonication followed by chromatin immunoprecipitation,X-ChIP)和非交联染色质免疫 共 沉 淀(native chromatin immunoprecipitation,N-ChIP)[6-7]。甲醛能交联固定蛋白与DNA,X-ChIP通过甲醛交联与超声处理形成可溶性染色质片段,利用特异性抗体沉淀目的蛋白与DNA复合物,通过解交联、蛋白酶消化等分离纯化DNA,常用于检测转录因子与DNA的相互作用[7]。微球菌核酸酶(micrococcal nuclease,MNase)兼具核酸内切酶和核酸外切酶活性,主要作用于核小体之间的连接DNA(linker DNA)。N-ChIP一般无需甲醛交联,MNase使染色质在连接DNA区域断裂,形成以核小体为单元的染色质片段,酶切后采用组蛋白修饰特异性抗体进行免疫共沉淀并分离纯化DNA,常用于组蛋白修饰的检测[8-9]。
通过超声或酶切断裂的DNA带有凸出的粘性末端,为连接具有平末端的测序接头,在构建文库过程中,需要对DNA进行末端修复。延伸DNA 3'端直至与5'端平齐,随后对5'和3'末端分别进行磷酸化和加dA修饰,获得具有5'磷酸化和3' dA的DNA片段,进一步连接测序接头[4,10]。图1显示了ChIP-seq基本流程:通过超声或酶切进行染色质片段化,利用特异性抗体免疫共沉淀目的蛋白和DNA,分离纯化的DNA经末端修复、连接测序接头,通过PCR扩增构建文库、测序并分析[11]。
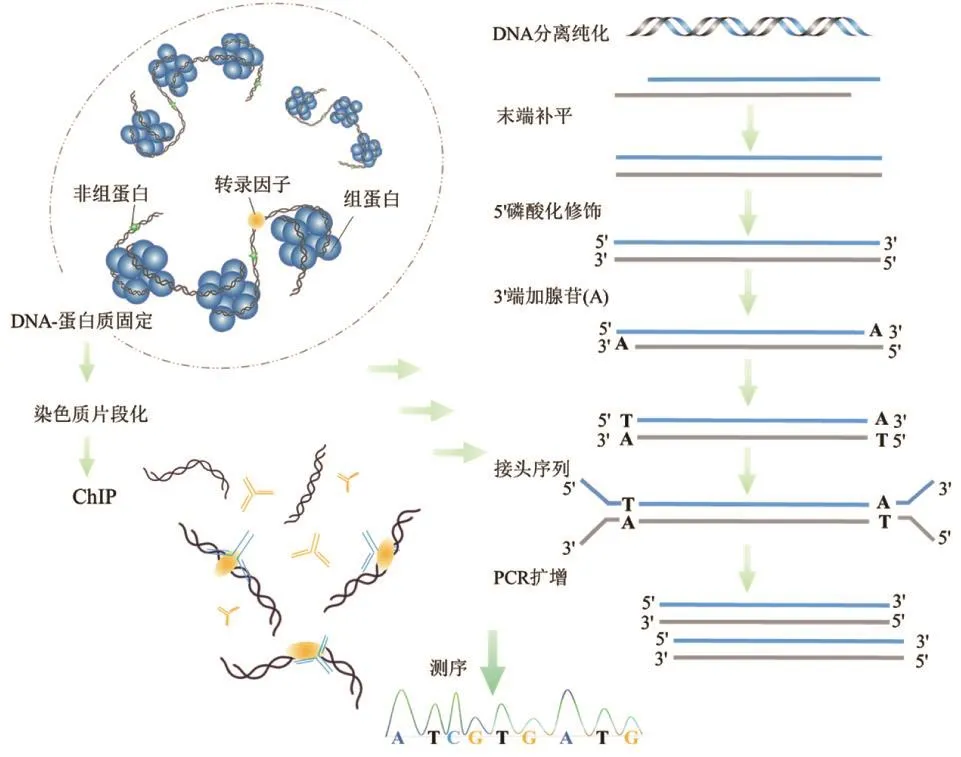
图1 ChIP-seq基本流程Fig.1 Major steps of ChIP-seq
1.2 ChIP-seq数据分析
ChIP-seq数据分析是通过将测序序列比对到参考基因组,识别富集区域内具有显著信号的峰、定位目的蛋白结合位点[12-13]。ChIP-seq数据分析基本流程包括:预处理及质量控制、序列比对、峰识别、可视化及高级分析等[14]。研究人员开发了多种基于不同算法的软件工具对高通量测序数据进行处理。其中,Linux系统的Conda和基于R语言的Bioconductor是开放式软件平台,具有ChIP-seq数据分析的大量工具和软件包[15]。
1.2.1 ChIP-seq数据分析软件及算法 测序数据质量控制包括质量评估和预处理。测序中,每个碱基具有一个质量值Q-score(Q-score= -log10e,e为测序错误率),用于衡量测序准确度,如Q30表明碱基识别发生错误的概率为0.1%,Q20指碱基识别发生错误的概率为1%。通过FastQC软件对测序的原始读长(raw reads)进行质量评估,一般使用质量值大于20或30的碱基占总体碱基的百分比(Q20或Q30)来评估测序数据质量。接头序列、扩增不均匀等将影响测序数据质量,这些情况可利用FastQC查看[16]。根据质量评估结果,可选择TrimGalore、Picard以及SAMTools等工具进行预处理,去除低质量碱基、接头序列以及PCR扩增重复(PCR duplicates)等,获得较高质量的数据(clean data)[17]。
序列比对通过将测序序列比对到参考基因组(或序列已知的基因组)进行定位[18]。高通量测序将产生大量的短序列数据,包含许多重复序列。绝大多数的序列比对算法构建索引数据库,通过索引筛选短序列在基因组中候选位置,减少搜索空间,提高比对效率[19]。根据建立索引数据结构方法的不同,短序列比对软件主要分为两类:基于哈希表(Hash table)数据结构和基于BWT压缩算法的索引数据结构(Burrows Wheeler transform,BWT)[20-22]。 哈 希表数据结构序列比对过程中,测序读段(reads)将以种子序列(seed)为单元生成序列集合,排列种子序列并建立索引数据结构进行比对[14,23]。该方法也可以对参考基因组生成种子序列,建立索引数据结构。BWT算法通过扫描短序列识别碱基重复的序列,将重复序列排列在一起,进一步压缩索引数据结构并重排列,以利于快速搜索和比对。目前,较为常用的短序列比对软件有Bowtie2、BWA、SOAP2和MAQ,不同的比对软件在比对数目、运行时间、内存消耗等方面各具优势和不足[24]。较短读长为单元时,可能的匹配区域很多,种子序列位点定位效率将降低,在进行序列比对过程中,基于Hash table数据结构的MAQ难以实现准确比对;基于BWT算法的比对工具有Bowtie2、BWA、SOAP2等,该算法的重排列利于短读长在基因组中候选位点进行快速搜索和比对[25]。测序碱基的深度与基因组覆盖率成正比例相关,随测序深度增加,基因组覆盖率增加,数据量更大。在HPV全基因组测序数据比对分析中,研究者采用上述4种工具将部分HPV测序数据与已知HPV基因组数据库进行比对,结果表明BWT算法的比对效率和计算速度优于Hash table算法[26]。
ChIP-seq数据分析中的重要环节是峰识别(peak calling)。峰(peak)被定义为基因组上reads富集的区域,峰识别是通过扫描比对到基因组上的短序列数据,进行样本数据和对照组数据的比较,识别富集区域[14]。MACS算法通过滑动窗口(sliding window)扫描,基于泊松分布模型统计显著的峰[27]。MACS采用具有固定大小的滑动窗口移动,可能产生窗口边缘识别模糊的问题。QuEST算法基于连续覆盖扫描,通过高斯核密度函数对reads富集密度进行评估,其中reads所在位置为窗口中心,具有最高密度值[28-29]。MACS通过计算全基因组范围内每个检测峰(peak)显著性P值,进一步分析差异peak,鉴定具有统计显著性的差异蛋白质结合位点,为较常用的峰识别工具[29]。
基于不同的需求,后续的数据处理和分析方法有所不同,如DNA序列特征的Motif分析、目的蛋白结合位点在基因组不同区域的偏好性分析、预测结合位点关联基因功能的GO注释及预测基因调控通路的Pathway分析等[30-31]。图2显示ChIP-seq数据分析流程及软件。

图2 ChIP-seq数据分析流程及软件Fig.2 Protocol for computational analysis of ChIP-seq data and software
1.2.2 ChIP-seq常见数据格式及可视化 数据格式对于合理组织数据存储,有效降低存储空间以及加快下游分析速度至关重要。图3显示了ChIP-seq数据常见格式、格式转变软件及可视化方法。fasta和fastq格式是存储核酸序列的常用格式,为二进制文本。其中,fastq格式包含短读序列和质量分数等信息[32]。为方便后续分析,可利用sratoolkit软件将测序原始数据格式转换为标准的fastq格式。SAM和BAM格式专用于存储参考序列的比对序列,是由基因组序列比对得到的输出格式[33]。BAM数据格式具有索引功能,为SAM格式的二进制,通过SAMTools软件可将SAM文件转换为BAM,有利于降低储存空间。UCSC基因浏览器可以读取BAM格式的数据,实现快速浏览[34]。储存转录因子、组蛋白修饰等结合位点在全基因组上的信号分布情况的数据格式为BedGraph(Bed)格式。该格式包含有染色体名称、染色体起始位点以及检出信号值。将MACS软件输出Bed格式文件转化为bigwig文件,上传到UCSC或IGV浏览器以实现数据可视化[35-36]。其他可视化工具有基于R语言的ChIPseeker和基于Python的deepTools等。
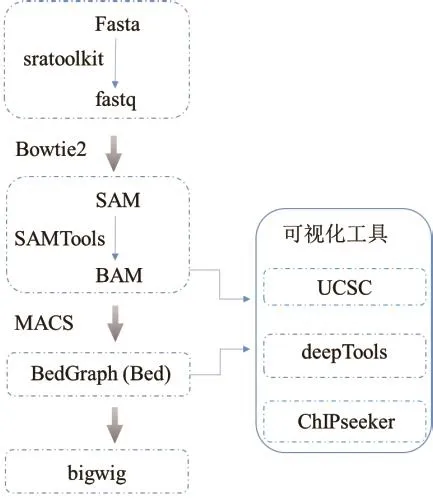
图3 ChIP-seq数据格式及可视化Fig.3 Standard data formats and visualization tools for ChIP-seq
2 基于ChIP-seq的优化技术
ChIP-seq需要大量细胞,面对稀少样本,收集足够多的细胞存在困难。免疫共沉淀过程,可能受到甲醛交联,非特异性结合的影响[37]。DNA片段中GC含量过高或过低,将导致PCR扩增偏倚,影响测序质量[38]。近年来研究人员针对上述问题进行优化并提出相应的技术。
2.1 基于细胞分离优化的ChIP-seq技术
流式细胞分选(fluorescence activated cell sorting,FACS)是利用鞘液包裹细胞形成样品流,通过流式细胞仪检测细胞携带的荧光信号,由分选器将特定的细胞从样本中分离出来。Amour等[39]通过FACS将细胞分离到含有裂解缓冲液的反应池中,进行细胞核分离与MNase酶切,提出基于MNase酶切的非交联免疫共沉淀ChIP-seq技术(nultra low input micrococcal nuclease-based native ChIP,ULI-NChIP),该技术适用于微量样品建库测序。该方法利用连续装置简化实验操作流程、减少分离纯化过程的洗涤次数,降低样品损失,利于在少量细胞中进行ChIP-seq 实验[40]。
微流控芯片通过产生非连续的液滴包裹单个细胞,实现单细胞分离[41]。Rotem等[42]基于液滴微流控芯片建立scChIP-seq(single-cell ChIP-seq):在具有多通道结构的芯片上,细胞裂解缓冲液包裹着标签序列与单细胞悬液汇聚并通过油相,形成“油包水”液滴,液滴内发生细胞裂解反应,随后与含MNase的凝胶微珠融合进行染色质片段化,进一步对数千个单细胞独立建库。scChIP-seq具有高度集成化、自动化优势,但微流控芯片使用成本较高,且微流控液滴操作对实验人员有较高技术要求。
2.2 基于酶切优化的ChIP-seq技术
lambda核酸外切酶、RecJf核酸外切酶具有5'-3'外切酶活性,分别作用于双链DNA和单链DNA,水解核苷酸之间的磷酸二酯键。Ho和Pugh将两种核酸外切酶引入到X-ChIP实验,提出ChIP-exo(ChIP combined with lambda exonuclease digestion)[43-44]。在特异性抗体沉淀目的蛋白与DNA交联复合物之后,通过lambda核酸外切酶消化,使双链DNA断裂末端最大程度的靠近蛋白,减少背景干扰,提高蛋白质结合区域分析的准确性。进一步解交联、分离纯化DNA,利用RecJf核酸外切酶消化单链DNA,减少背景噪音。ChIP-exo核酸外切酶处理,有利于提高检测转录因子结合位点的分辨率[2,44]。
2.3 基于免疫共沉淀优化的ChIP-seq技术
染色质免疫共沉淀实验过程中,通常使用磁珠富集抗体、蛋白和DNA复合物,但难以避免非特异性结合的影响[37]。Zhu等[45]基于微流控芯片优化ChIP实验操作流程,提出MOW ChIP-seq(microfluidic oscillatory washing-based ChIP-seq)。将超声处理的染色质载入芯片,采用偶联有目的蛋白特异性抗体的磁珠富集可溶性片段。通过控制芯片上微量流道内样品与磁珠的流动速度,促进两者混合均匀,提高免疫磁珠的捕获效率。另外利用微流控振荡辅助,降低非特异性结合,提高结果准确度。MOW ChIP-seq通过结合免疫磁珠、微流控芯片对样品进行分离与富集,具有试剂和样品消耗量少、自动化等优势[37]。
2.4 基于测序文库优化的ChIP-seq技术
ChIP-exo增加的酶切步骤以及多次洗涤,使用于建库的起始DNA量减少,需要增加PCR循环扩增数,可能导致过多的重复序列[46]。在ChIP-exo基础上,He等[47]设计含特异性序列的测序接头,通过DNA自环化(self-circularization)方法优化建库,提 出 ChIP-nexus(ChIP experiments with nucleotide resolution through exonuclease,unique barcode and single ligation)。建库过程中,将带有限制性内切酶BamH I酶切位点序列的测序接头连接到DNA,利用环化连接酶将DNA自身环化。环化DNA通过BamH I酶切处理,其断裂产物两端将带有测序接头,利于直接扩增建库。ChIP-nexus通过分子内自身环化进行接头连接构建文库,其连接效率比ChIP-seq中DNA连接酶的连接效率更高,有利于降低建库DNA需求量。
扩增构建测序文库过程中,高GC含量DNA片段易产生扩增偏倚,扩增体系中的引物二聚体将造成背景信号[48]。Adli等[49]对文库构建方案优化,并提出Nano-ChIP-seq。在PCR扩增建库过程中,使用发夹结构引物,减少引物二聚体。针对高GC含量序列,使用Phusion高保真DNA聚合酶,并优化缓冲液和扩增循环次数。此外,在引物序列中引入限制性内切酶BciV I的酶切位点序列,扩增产物在内切酶作用下产生3'A突出末端,利于直接连接测序接头。研究表明,利用Nano-ChIP-seq建库方案可从少量细胞中进行ChIP-seq检测[49-50]。表1比较了上述ChIP-seq优化技术。

表1 ChIP-seq优化技术比较Table 1 Comparison of optimization techniques of ChIP-seq
3 CUT&RUN、CUT&Tag及CoBATCH技术
ChIP包含细胞裂解、细胞核提取、染色质制备以及免疫共沉淀等多个连续步骤,需要的细胞量较大,常规ChIP-seq很难进行少量细胞或单细胞水平的检测[51]。最近,研究人员利用目的蛋白特异性抗体使MNase或Tn5转座酶“靶向”作用于目的蛋白结合位点附近的染色质,提出CUT&RUN、CUT&Tag等方法。与ChIP-seq相比较,这些方法无需获取可溶性染色质进行免疫共沉淀,具有实验流程简单、耗时短等优势,可以在少量细胞或单细胞水平进行检测。
3.1 CUT&RUN技术
Schmid等[52]建立了以酶“靶向”切割染色质的 方 法:ChIC(chromatin immunocleavage)。 为 使MNase选择性地作用于目的蛋白结合区域,研究者利用对抗体具有亲和力的Protein A融合MNase(pAMN),通过抗体结合pA-MN融合蛋白将MNase间接结合到目的蛋白。在ChIC实验中,首先利用螯合剂EDTA、EGTA等抑制MNase酶活性,将细胞与含有目的蛋白特异性抗体、pA-MN的缓冲液反应后,用Ca2+激活MNase,使其在特定位点断裂DNA。2016年,Skene等[51]将ChIC与高通量测序技术结合,提出CUT&RUN。
CUT&RUN中,特异性抗体先与目的蛋白结合,进一步与pA-MN反应,募集pA-MN到结合位点。在0℃下保持较低的酶活性,抗体固定酶后,采用Ca2+激活MNase使其在目的蛋白结合位点附近作用于染色质开放区域,酶切后染色质片段释放,进一步建库测序[51,53]。研究者利用伴刀豆球蛋白A(concanavalin A,Con A)能与细胞的多糖、糖蛋白特异结合特性,将Con A包被在磁珠表面,固定细胞或细胞核,以利于洗涤。2018年,Skene等[54]对CUT&RUN实验优化,通过洋地黄皂苷(digitonin)增加细胞膜通透性,释放酶切染色质片段到细胞外,由于未被切割的染色质仍留在细胞内,有利于降低背景噪音。2019年,Hainer等[55]利用流式细胞分选将单个细胞分离到多孔板中进行CUT&RUN实验,提出可对极少量细胞进行检测的ULI-CUT&RUN(ultra-low input CUT&RUN)。
3.2 CUT&Tag及CoBATCH技术
Tn5转座子由核心序列和两末端序列组成。Tn5转座酶可以与Tn5转座子的末端序列结合形成复合物,该复合物具有“剪切-粘贴”(cut and paste)催化活性,两者协同完成Tn5转座子末端序列的切割和转移。在Tn5转座复合体作用下,转座子末端DNA的磷酸二酯键被水解并产生3'- OH羟基末端,随后Tn5转座酶进攻目标DNA形成9 bp的切口,同时转座子羟基末端与目标DNA的磷酸基团之间形成共价键,从而将转座子序列插入到目标DNA[56]。由于这一特性,Tn5转座酶可将测序接头序列随机插入染色质开放区域,进行DNA片段化和测序接头连接[57]。Schmidl等[58]首先将 Tn5转座酶工具与ChIP相结合,代替ChIP-seq构建测序文库中的末端补平、3'末端加A等处理,提出ChIPmentation。其中,测序接头和Tn5转座酶组装形成转座复合物,与X-ChIP获得的目的蛋白结合DNA反应,进行接头连接。
2019年,Kaya-Okur等[59]参考 CUT&RUN的实验流程,将带有测序接头的Tn5转座酶与Protein A融合(pA-Tn5),利用pA-Tn5替换pA-MN,提出CUT&Tag。特异性抗体结合目的蛋白后,进一步与pA-Tn5反应,利用Mg2+激活Tn5转座酶活性进行染色质切割与接头连接。Tn5转座酶建库有利于减少损失,与CUT&RUN比较,CUT&Tag的样本需求量更小。2020年,Bartosovic等[60]将液滴微流控技术与CUT&Tag相结合,进行单细胞建库测序,提出 scCUT&Tag(single-cell Cut&Tag)。 表2比 较 了CUT&RUN、CUT&Tag与 ChIP-seq。

表2 CUT&RUN、CUT&Tag与ChIP-seq比较Table 2 Comparison of CUT&RUN,CUT&Tag and ChIP-seq
标签组合(combinatorial indexing)建库是利用不同的barcode序列为不同样品的DNA进行组合标签标记,通过一次建库可区别成千上万单细胞,利于提高单细胞测序的通量,获得更多单细胞信息[61]。2019年,Wang等[62]基于标签组合和pA-Tn5提出 CoBATCH(combinatorial barcoding and targeted chromatin release)。通过流式细胞仪和微孔板进行单个细胞的分选和分离,采用带有T5/T7组合标签的pA-Tn5对染色质进行转座酶切割和标签标记,进一步对带有不同标签的DNA建库与测序。CoBATCH利用组合标签区分不同样本来源的细胞,同时组合标签增加了测序文库的复杂度,利于进行高通量的单细胞检测。
4 在植物基因表达调控研究中的应用
研究人员使用ChIP-seq检测植物转录因子结合位点、组蛋白修饰分布,已广泛应用于生物钟调控、激素信号转导、光信号途径、胁迫响应等研究[63-65]。CUT&RUN和CUT&Tag方法具有流程简单、良好的可重复性、需要的细胞数量少等优势,近年来,被初步应用于植物转录因子、组蛋白修饰H3K27me3和 H3K4me3 等研究[66-68]。
4.1 植物转录因子结合位点相关研究
植物的生长发育除了受自身遗传因素的调控外,还受到环境胁迫、内源激素变化等影响。转录因子在植物的生物钟调控、激素信号转导、生长和代谢等过程中发挥重要作用[69-70]。GRF7(growthregulating factor 7)是水稻(Oryza sativa)生长调节因子类转录因子,Chen等[71]选用不同发育时期的水稻幼穗进行ChIP-seq检测,发现OsGRF7与细胞色素P450基因OsCYP714B1和生长素响应基因OsARF12启动子中的ACRGDA motif结合,激活基因转录,参与赤霉素的合成和生长素的信号传导途径,调控幼穗发育。生物钟是影响生物昼夜节律的重要因素,CAA1(circadian clock-associated 1)是拟南芥(Arabidopsis thaliana)生物钟重要转录因子,PRRs家族基因参与生物钟调控,Kamioka等[69]通过ChIP-seq发现CCA1直接结合在基因PRR5的启动子区域,抑制PRR5表达,并发现CCA1与PRR9、PRR7、PRR5等基因启动子上多个motif结合,包括G-box、EEs、CT重复、TCP等,调控生物钟周期。光是调控植物生长和发育的重要环境因素,FHY3(far-red elongated hypocotyl 3)是拟南芥光信号转录因子,Ouyang等[72]利用ChIP-seq在远红光条件下鉴定到FHY3结合在基因FHY1和ELF4启动子的FBS motif(CACGCGC),激活基因表达,促进光敏色素A在细胞核的积累,进而调控光信号途径;并发现FHY3与叶绿体分裂相关基因ARC5启动子区域的FBS motif结合,激活其转录,进而影响叶绿体发育。叶夹角是影响株型与作物产量的重要农艺性状,油菜素内酯(brassinosteroid,BR)是植物重要的促生长类激素。BR促进细胞伸长与分裂,对水稻叶夹角发育有影响,Guo等[68]通过CUT&RUN分析发现,水稻bHLH(basic-helix-loop-helix)转录因子家族的OsbHLH98,结合水稻BR信号转导途径相关基因BUL1的启动子上的G-box、E-box等motif,抑制基因表达,调控水稻叶夹角发育。
4.2 植物组蛋白修饰及分布相关研究
在植物发育过程中,除了转录因子激活或抑制基因表达的作用外,染色质组蛋白翻译后修饰通过影响染色质结构来调控基因的表达。组蛋白修饰H3K27me3抑制基因转录,Wu等[73]通过ChIP-seq发现在高氮素条件下水稻分蘖抑制基因D14和OsSPL14启动子区域的H3K27me3富集水平显著升高。进一步研究表明,水稻分蘖期氮素应答关键蛋白NGR5通过与PRC2相互作用,将PRC2招募到D14和OsSPL14的启动子上催化H3K27me3修饰并抑制基因表达,进而调控氮浓度对水稻氮素吸收和分蘖发生的影响。Nishio等[74]以鼠耳芥(Arabidopsis halleri)为材料,通过ChIP-seq分析组蛋白修饰H3K27me3对其季节性和昼夜节律基因表达的影响,并与组蛋白修饰H3K4me3进行比较。发现H3K27me3具有季节性的可塑性与昼夜节律稳定性,H3K27me3的信号变化晚于H3K4me3出现,在环境变化中进行长期的基因表达调控。
研究者将CUT&RUN、CUT&Tag用于植物组蛋白修饰分析,并与ChIP-seq进行比较。Zheng等[66]利用流式细胞仪分选拟南芥胚乳细胞核,通过CUT&RUN分析发现胚乳细胞周期中有丝分裂间期的相关基因被H3K27me3修饰,H3K27me3影响亲本等位基因的差异表达与胚乳发育。与ChIP-seq相比较,CUT&RUN检测所需细胞数量更少。Tao等[67]将CUT&Tag用于对棉纤维细胞基因组及外显子、内含子、启动子等区域组蛋白修饰H3K4me3的分布特征分析,发现H3K4me3显著富集在基因启动子上(转录起始位点上游1-2 kb)。使用相同数量细胞进行CUT&Tag和ChIP-seq检测,发现CUT&Tag分析结果具有较好重复性,显示出更高分辨率和更低的背景信号,所需实验时间更短。
5 总结与展望
ChIP-seq已被广泛用于转录因子和组蛋白修饰研究。近年来,研究人员将流式细胞分选、微流控芯片与ChIP-seq相结合,优化细胞分离、染色质片段化、免疫共沉淀以及测序文库构建等关键步骤并提出优化方法。CUT&RUN、CUT&Tag利用“靶向”酶切和Tn5转座酶建库,简化实验流程。在上述基础上,使得ChIP-seq在少量细胞或在单细胞水平的检测成为可能。其他测序技术在转录因子和组蛋白修饰研究中具有重要作用:微球菌核酸酶测序MNase-seq(micrococcal nuclease sequencing) 利 用MNase切割染色质,获取核小体DNA建库测序,绘制核小体定位图谱[75]。MNase-seq与ChIP-seq联合将利于分析目的蛋白结合位点附近的核小体定位状态。染色质转座酶可及性测序ATAC-seq(assay for transposase-accessible chromatin with high throughput sequencing)利用Tn5转座酶进行染色质切割与接头连接,检测染色质开放区域[76]。ATAC-seq结合ChIP-seq可进一步分析转录因子、组蛋白修饰对染色质开放性的影响。ChIP-seq与转录组测序技术(RNA-seq)的联合分析,有利于进一步确认转录因子以及组蛋白的修饰对于基因表达的调控作用。
随着技术的不断发展,ATAC-seq、RNA-seq,CUT&RUN、CUT&Tag等均可以实现单细胞水平的检测。进行单细胞的组学分析,克服细胞异质性对有效信号的“干扰”问题,有利于珍稀样品的准确分析。将ChIP-seq纳入多组学联合分析,帮助更全面的理解细胞内蛋白与DNA相互作用,具有重要的研究意义。
