机器学习加速搜寻新型双钙钛矿氧化物光催化剂*
2022-09-14万新阳章烨辉陆帅华吴艺蕾周跫桦王金兰
万新阳 章烨辉 陆帅华 吴艺蕾 周跫桦 王金兰
(东南大学物理学院,南京 211189)
A2BB′O6 型双钙钛矿氧化物材料,相比于ABO3 型单钙钛矿氧化物材料,具有更好的稳定性和更宽泛的能带选择范围,在光催化全解水领域具有良好的应用前景.然而,由于晶体结构和组成元素的多样性,实验和理论上快速、准确搜寻高催化活性的A2BB′O6 型双钙钛矿氧化物材料具有相当大的挑战性.本文由材料数据库的带隙值数据出发,采用机器学习与第一性原理相结合的方法,从50000 多种A2BB′O6 型双钙钛矿氧化物材料中筛选出近8000 种可能适用于光催化全解水的材料.对筛选结果的统计分析表明,B/B′位均为d10 金属离子的双钙钛矿氧化物,更有可能成为全解水光催化剂.随后通过进一步的第一性原理计算挑选出Sr2GaSbO6,Sr2InSbO6 和K2NbTaO6 这3 种带边位置合适且不含铅、汞离子的A2BB′O6 型双钙钛矿氧化物材料作为候选的全解水光催化剂.
1 引言
如何减少化石燃料的使用以及降低二氧化碳的排放,并寻找可持续的清洁能源,一直都是人们广泛关注的问题[1-3].太阳能是目前最直接、最广泛的清洁能源,如何将光能有效地转化为电能或化学能[4,5]是寻找清洁能源的关键,而光催化全解水被认为是最理想、最方便的手段[6-8].在高效光催化全解水反应中,光催化剂的选取则是光催化反应的核心.相比于价格昂贵、含量稀少的贵金属催化剂,钙钛矿氧化物材料因其结构稳定、种类多样、易合成、价格低廉等优点近些年受到大家广泛的关注[9-11].然而ABO3型单钙钛矿材料普遍带隙值过大,导致其对可见光的吸收效率较低.最近,新型A2BB′O6双钙钛矿氧化物在实验上被成功合成并展现出更好的稳定性和光催化活性[12,13].在A2BB′O6双钙钛矿氧化物中,B/B′位通常由高价元素组成,其组分相较于单钙钛矿更为复杂多样[14],同时结构中的BO6与B′O6八面体结构交替排列,形成的B-O-B′结构产生超交换作用[15],影响其能带结构,使其具有更宽泛的带隙选择范围.因此,A2BB′O6型氧化物双钙钛矿材料被认为能够更好地满足光催化全解水的要求,有着很广阔的应用前景.
然而,A2BB′O6型双钙钛矿氧化物多样的元素组合在带来多样化的结构和性质的同时,也对从不同组合中找到适用于光催化全解水的候选材料造成了不小的困难.基于第一性原理计算的材料设计方法在钙钛矿材料设计中具有不错的准确性,但对104数量级的搜索空间来说,依然需要一种更快速的筛选方法.随着材料数据库的不断完善,机器学习作为一种数据驱动的方法[16],能够在极短时间内对大量材料进行快速筛选,已经在新材料设计中展现出了巨大的潜力,例如二维铁磁材料[17,18]、双钙钛矿卤化物光伏材料[19]、多相催化剂[20]等.此外,传统的机器学习模型虽然能够比较成功地预测出新型材料,但往往局限于某一单一体系,同时对预测材料之间关联性的挖掘也不足.因此,提升机器学习模型对不同结构的适用性、挖掘目标材料的构效关系也是该领域研究关注的重点问题.
本工作的整体研究流程如图1 所示.通过机器学习和第一性原理结合的方法,构建了新型A2BB′O6型双钙钛矿氧化物(后文简称双钙钛矿)材料快速筛选的框架,包含数据准备、特征选择、带隙分类模型和回归模型的构建,以及对预测结果构效关系的统计分析和计算结果验证.首先,通过整合钙钛矿材料数据库和高通量计算,获得了近2500 种钙钛矿氧化物材料数据.其次,建立了两步机器学习模型框架,第一步通过机器学习分类模型,挑选出带隙值大于0.2 eV 的双钙钛矿材料,第二步建立机器学习回归模型,对分类模型结果中带隙大于0.2 eV 的双钙钛矿材料进行预测,得到准确带隙值.然后,将机器学习模型用于预测集,对50000 多组新双钙钛矿材料带隙进行了快速预测和筛选,得到近8000 种新型光催化材料.在此基础上,进一步对预测结果进行统计分析,总结了B/B′位金属离子组成以及钙钛矿结构的对称性对双钙钛矿带隙大小的影响规律.最后,挑选了29 个带隙值在0.2 eV 以上的双钙钛矿材料进行DFT 的验证,并最终挑选出3 种不含铅汞离子、带隙大小合适、带边位置合适的双钙钛矿材料作为全解水光催化剂.
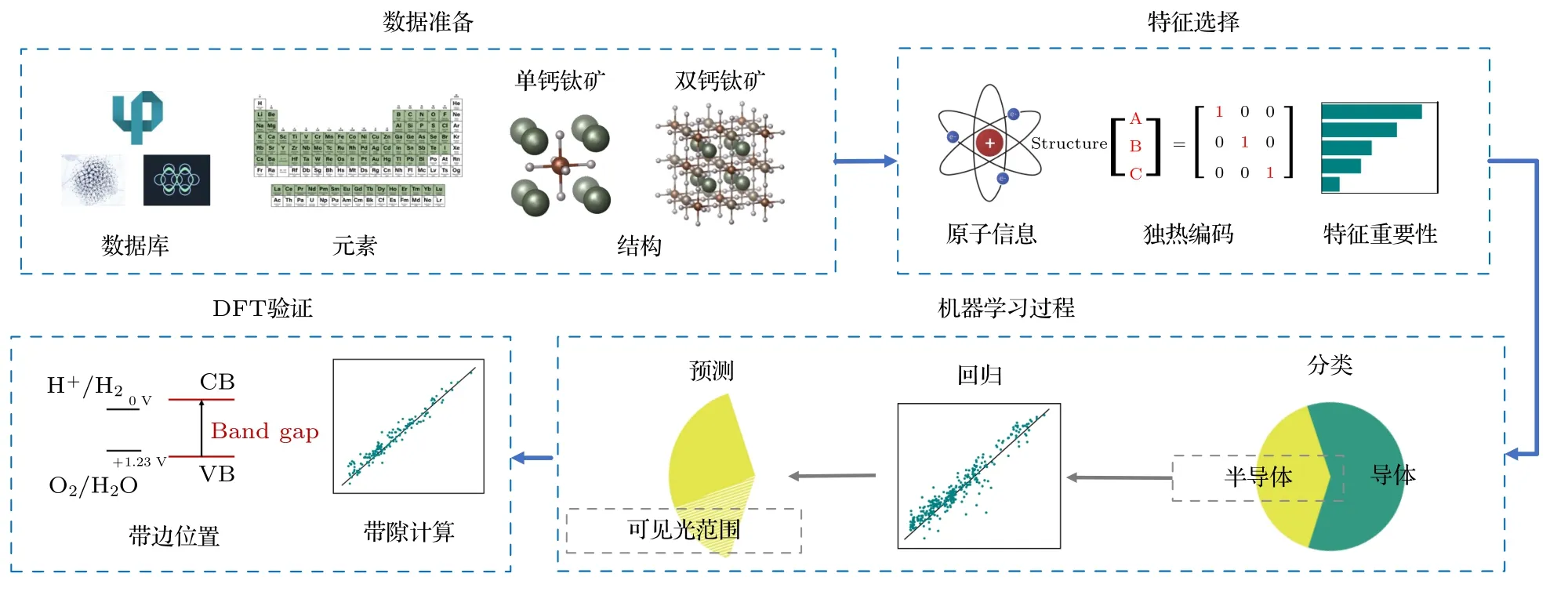
图1 基于两步机器学习算法的双钙钛矿氧化物筛选框架.包括数据准备、特征选择、机器学习过程和DFT 验证4 个步骤Fig.1.Multistep machine learning-based screening framework for double perovskite oxides.There are four steps including data collection,feature selection,machine learning process and DFT verification.
2 计算方法
2.1 XGBoost 算法
本文使用XGBoost (eXtreme gradient boosting)[21]算法训练分类和回归模型.不同于梯度提升(gradient boosting)[22]算法,XGBoost 算法在损失函数上使用了二阶泰勒近似,使得其与Newton Raphson 方法相联系.
2.2 密度泛函理论计算
本文采用基于密度泛函理论(density functional theory,DFT)的第一性原理计算方法,通过应用软件VASP (Vienna ab-initio simulation package,VASP)[23]来计算材料的电子结构.计算采用了广义梯度近似(generalized gradient approximation,GGA)下的PBE (Perdew-Burke-Ernzerhof)泛函[24],并考虑了铁磁与A 型反铁磁的情况[25].采用投影缀加平面波(projected augmented wave,PAW)方法来描述离子实和价电子之间的相互作用[26].平面波的截断能设置为500 eV,并采用G中心的Monk horst-Pack 方法[27]进行第一布里渊区采样,k点网格选取为 3×3×3,离子弛豫的能量收敛标准设置为10—6eV,原子之间相互作用力的收敛标准为0.02 eV/Å.对于过渡金属元素,参考Materials Project 的数据来源,引入了相应的Hubbard U[28]修正进行相关计算,修正值如附录表A1 所示.对于最终预测的3 个新型双钙钛矿材料,采用HSE06(hybrid Heyd-Scuseria-Ernzerhof,HSE)杂化泛函[29]进行了进一步的计算.

表A1 过渡金属计算时附加的Hubbard U 值Table A1. Hubbard U value for the transition metal elements.
3 结果讨论
3.1 数据准备
首先,从AFLOW[30],OQMD (the open quantum materials database)[31]和Materials Project[32]等常用数据库中挑选出钙钛矿结构的氧化物数据.单钙钛矿氧化物和双钙钛矿氧化物的化学式分别表示为ABO3和A2BB′O6,但并非所有满足这一元素组成和化学式的材料都是钙钛矿结构.经典的钙钛矿结构判定方式是Goldschmidt 提出的容忍因子t[33],然而这种判定方式原本是基于单钙钛矿材料提出的,且在很多情况下不够准确.很多研究团队因此对钙钛矿判定条件进行了更进一步的研究,如Sun 等[34]提出的 (µ+t)η,Bartel 等[35]提出的新容忍因子,Weng等[36]提出的µ/t等.考虑分类准确率,采用了Filip等[37]提出的基于八面体、倾斜度等限制的多重判定条件.利用该判定条件对前述材料数据库进行数据清洗,并通过结构相似度分析[38]剔除重复数据后,得到单钙钛矿(ABO3)1027 组和双钙钛矿(A2BB′O6) 463 组.根据结构相似度分类结果,保留主要的3 种类别,分别命名为“立方类结构”“正交类结构”和“菱方类结构”,结构如图2(a)所示.
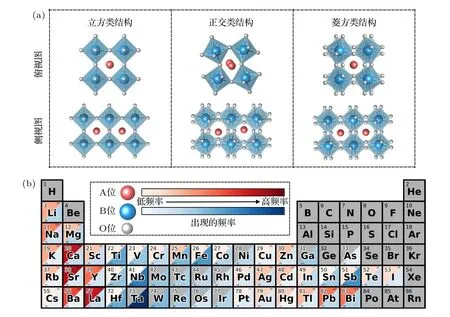
图2 (a) 3 种不同的钙钛矿构型;(b) 训练集中A 位和B 位元素出现的频率Fig.2.(a) Three different perovskite structures;(b) occurrence frequency of A-and B-site elements in the training set.
众所周知,机器学习模型构建需要足够多的数据量作为支撑.根据以上筛选方法得到的463 组双钙钛矿数据较少,为了保证训练数据的完备性,将数据库中满足判定条件的1027 组单钙钛矿数据加入训练集中(等价于B′位与B位相同的双钙钛矿).同时,从立方类结构的双钙钛矿(结构简单,便于大量获取数据)中随机挑选数据库中没有的1005 组进行第一性原理高通量计算,得到相应的PBE 带隙值.在双钙钛矿结构中,会因为等效的B位和B′位阳离子不同的排布方式,对带隙产生不同的影响.在本工作的的数据中并未对每种结构不同的排布一一计算,而采用最常见的F 型阳离子排布方式.其中计算数据皆为F 型排布,数据库主句中绝大部分为F 型排布,这样的排布类型能量往往要远低于其他排布类型[39].最终合计得到2495 组钙钛矿氧化物的带隙值,这足以支持机器学习模型训练[19,40].预测集则由满足钙钛矿结构判定条件,且并未在训练集中出现的双钙钛矿构成,共56894 组数据.从图1(b)中可以看出训练集双钙钛矿A位和B位(包含B′位)元素分布情况,即A位元素主要出现在碱金属、碱土金属、类金属及过渡金属区域的ⅠB,ⅡB 和ⅢB 副族,而B位元素主要集中在过渡金属区域.其原因在于B与B′位6 配位的环境决定了B位元素离子半径较小(过渡金属离子等),而A位离子半径较大(稀土或碱土金属元素).因此导致了钙钛矿结构中元素多变的价态,影响钙钛矿包括电子结构在内的多种性质[41].
除数据量外,影响机器学习模型准确性的另一个主要因素是特征(feature)与目标性质的关联性.为了综合考虑元素组分因素和晶体结构因素,本文选取构成双钙钛矿A位和B/B′位原子的物理性质(单钙钛矿B′位同B位)作为一部分特征,将3 种不同构型钙钛矿材料进行独热编码[42]作为另一部分特征,剔除信息冗余和与目标性质关联不高的特征后,最终获得45 个特征来组成描述材料的特征向量,特征符号表示如附录表A2 所示.
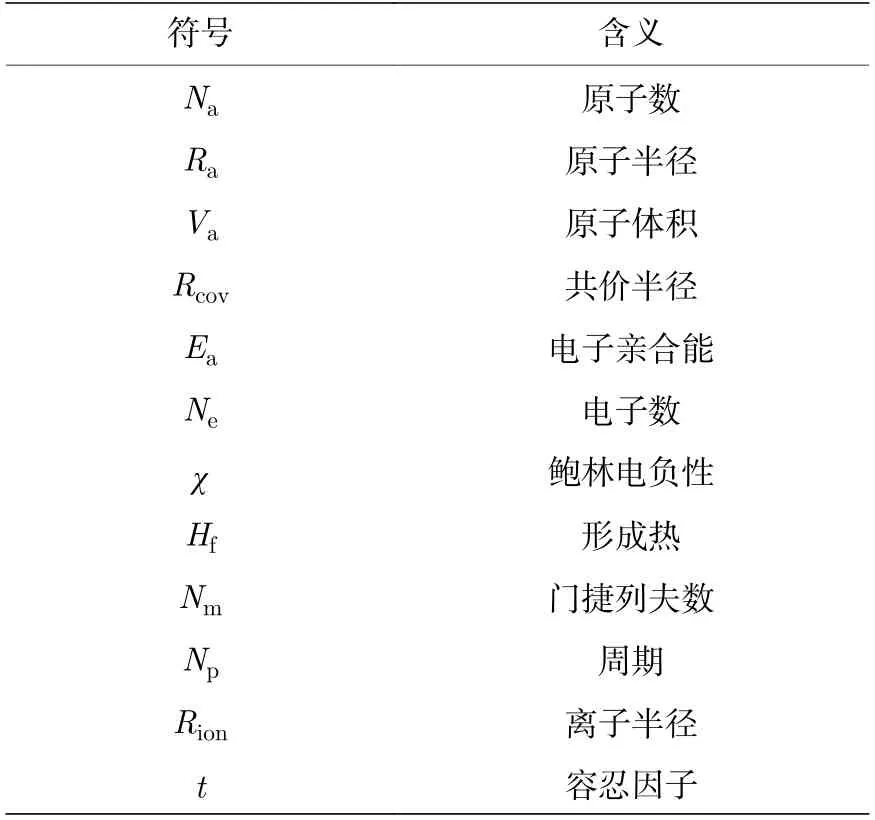
表A2 特征符号及含义Table A2. The symbol of features and their corresponding meanings.
3.2 机器学习模型对带隙的预测
为了提升双钙钛矿材料带隙预测的准确率,本文采用了两步建模的方法.第一步,选择适用于结构化数据与中低维度特征的XGBoost 算法训练一个分类模型,用来筛选PBE 带隙值大于0.2 eV 的钙钛矿材料,排除金属或光吸收明显不佳的窄带隙材料.在训练和测试集的数据中共有757 组数据的带隙值大于0.2 eV.图3(a)列出了特征重要性分析中排在前10 的特征,可以看出结构独热编码(structure)的重要性要远高于其他特征,意味着结构的对称性对钙钛矿材料的带隙值影响显著.图3(a)中分类结果的混淆矩阵表明分类模型特异度为94.6%,表明了该模型可以有效筛除0.2 eV 以下的双钙钛矿材料.机器学习分类模型性能常用受试者操作曲线线下面积 (area under curve,AUC)评估,其值一般在0.5—1.0 之间,越接近于1.0 模型效果越好.由图3(b)可以看出,带隙分类模型在测试集上的AUC 值可达0.86,证明了模型的可靠性.将训练好的分类模型应用到56894 组预测数据上,得到了14407 组带隙值大于0.2 eV 的双钙钛矿材料,带隙大于0.2 eV 的数据占预测数据总量的1/4 左右.分类过程作为第一步筛选,剔除了大量金属性或窄带隙的双钙钛矿材料,有助于回归模型的进一步构建.
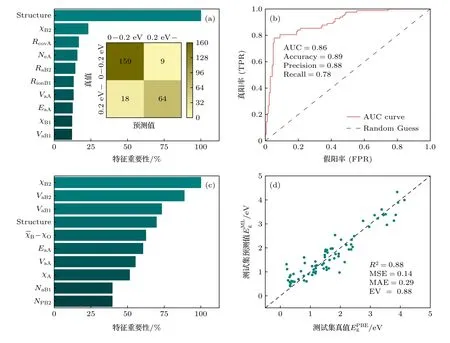
图3 (a) 分类模型中重要性前十的特征和混淆矩阵;(b)分类模型测试集ROC 曲线和AUC 值;(c)回归模型中重要性前十的特征;(d)回归模型测试集R2,均方误差,平均绝对误差和解释方差Fig.3.(a) Relative feature importance of top 10 most important features and confusion matrix for bandgap classification;(b) receiver operating characteristic (ROC) curve for bandgap classification test set,area under the ROC curve (AUC) is provided;(c) relative feature importance of top 10 most important features for bandgap regression;(d) performance of bandgap regression model,coefficient of determination (R2),mean square error (MSE),mean absolute error (MAE) and explained variance (EV) are provided.
第二步,为了得到准确的带隙值,继续利用XGBoost 算法来建立带隙值的回归模型.新的训练集和测试集是由第一步回归后带隙值大于0.2 eV的757 组钙钛矿构成,包括257 个ABO3单钙钛矿和500 个A2BB′O6双钙钛矿.从特征的重要性分析可知,B位原子的电负性、B位原子体积和结构为重要的特征(图3(c)).训练的回归模型在测试集上的准确度用决定系数R2表示,其值为0.88,均方误差MSE 为0.14 eV(图3(d)).这一结果说明此回归模型对于双钙钛矿氧化物带隙值的预测是可靠的.最后,利用训练好的回归模型应用到前一步得到的14407 个双钙钛矿氧化物体系,得到了其PBE 带隙值.值得注意的是,在 PBE 计算中,半导体氧化物的带隙,尤其是钙钛矿氧化物的带隙,相较实验值通常被低估了40% 到 50%[43,44].因此,基于PBE 数据预测的双钙钛矿氧化物材料带隙也会同样被低估.对于可见光能量范围内(1.6—3.1 eV)的光催化全解水反应,优先选择预测PBE 带隙在0.8—1.8 eV 的范围内的材料,共有7909 个,占总体预测数据的13.9%.这些双钙钛矿氧化物均有可能成为光催化全水解反应的催化剂.
3.3 带隙数据的统计分析
为了进一步分析组分和结构对双钙钛矿氧化物带隙的影响,对机器学习预测结果进行了统计分析.众所周知,具有八面体结构且含有d0和d10金属离子的氧化物被广泛应用于光催化全解水领域,原因在于此类氧化物材料的导带是由金属原子的d 轨道和sp 轨道构成,价带由O 的2p 轨道构成[45],因而改变金属原子可以有效地调节电子结构,进而提高催化活性.一般地,d0金属离子为: Ti4+,Zr4+,Nb5+,Ta5+,Mo6+和W6+;d10金属离子为: Ga3+,In3+,Sn4+和Sb5+[46-48].针对以上d0与d10金属离子,主要关注B或B′位离子为d0或d10电子构型的双钙钛矿材料.
首先,对比预测集中B/B′位为d0/d10金属离子与B/B′位离子不含d0/d10离子体系的带隙值是否存在区别.如图4(a)所示,带隙值越大,B/B′位离子含d0/d10离子的双钙钛矿占比越大,而B/B′位离子均不含d0/d10离子的双钙钛矿占比越小.在0.8—1.8 eV 范围内,B/B′位均不含d0/d10离子的体系占26%,B/B′位中有一个为d0/d10离子的体系占31%,而B/B′位都是d0/d10离子的体系占到43%.因而,对于可见光吸收能量范围对应的PBE 带隙值,双钙钛矿材料中有74%的B/B′位含有d0/d10离子.这进一步证实,B或B′位离子为d0或d10电子构型的双钙钛矿材料,更适合作为光催化候选材料.
进一步探究了B/B′位包含d0或d10离子的具体类别对回归模型预测带隙值的影响.如图4(b)所示,B/B′位如果只包含d10离子(蓝色),带隙值分布的峰值所对应的带隙最小,分布范围集中在0.5—2.0 eV 之间.对于B/B′位只包含d0离子(红色)的双钙钛矿,数量较少,其带隙值要稍大于B/B′位只包含d10离子的钙钛矿材料,带隙值分布主要集中在1.2—2.0 eV 之间.而B/B′位同时包含d0/d10离子(灰色)的双钙钛矿,数量最多,带隙值的分布范围也最广,涵盖了1 eV 以上的广泛区间.图4(b)中绿色区域为适合光催化的带隙范围,从带隙分布上来看,此区域主要包含了B/B′位只有d10离子(蓝色)的双钙钛矿材料与B/B′位只有d0离子(红色)的双钙钛矿材料.可知B/B′位只有d10离子(蓝色)或只有d0离子(红色)的双钙钛矿材料更有望成为带隙合适的光催化材料.
然而上述结论并未考虑分类模型筛除的双钙钛矿材料(带隙0—0.2 eV).因此,想要在实验上指导双钙钛矿B与B′位点元素该如何选择,还需综合分类与回归的预测结果,整体分析带隙的分布与B或B′位所包含离子的关系.在回归带隙统计分析的3 种情况之外,扩充选取B/B′位只有一个是d0或d10离子,另一个非d0或d10离子的两种情形,即B/B′位5 种情况下的带隙分布(图4(c)).从图4(c)可以看出,B/B′位中只有一个是d0或只有一个d10离子两种情况带隙分布类似,大部分数据集中在0.2 eV 带隙值以下,并不适合作为光催化候选材料.而在0.8—1.8 eV 的能量范围内B/B′位都为d10离子的占比最大,为37%,此带隙范围内B/B′位都为d0离子占比其次,达到23%.综合以上讨论,可以得出:B/B′位均为d10离子的双钙钛矿氧化物材料最适合做光催化全解水的候选材料,其次是B/B′位均为d0离子的体系.
钙钛矿结构对称性对材料的带隙值有何种影响也是值得关心的一个问题.从双钙钛矿材料回归模型预测的结果里挑选出B位为d0/d10离子的材料,查看对于同样的化学式,不同的晶型与带隙大小的对应关系.从图4(d) 中可以看出,低对称性的菱方类结构(蓝色)和正交类结构(红色)数据点整体位于高对称性的立方类结构(黑色),对比图1(a)中结构对称性结果,发现双钙钛矿材料的对称性越低,其带隙值越大.
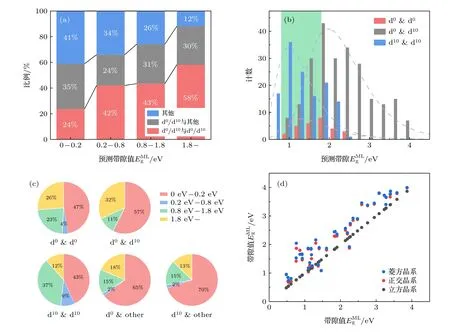
图4 (a) 预测的钙钛矿 带隙值百分比统计图,红色区域的代表 双钙钛矿的 B/B′ 位都是d0 或d10 金属离子,灰色区 域代表B/B′ 位点中只有一个是 d0 或 d10 金属离子,蓝色区域代表B/B′ 位点不含d0 或 d10 金属离子;(b) B/B′ 位点都是d0 或d10 金属离子的双钙钛矿带隙分布图,绿色区域为可见光能量范围;(c)不同B/B′ 位组分下双钙钛矿带隙统计图;(d)相同化学式下,3 种晶系结构的双钙钛矿带隙值,其中B/B′ 位都是d0 或d10 金属离子Fig.4.(a) The percentage chart of predict set of bandgap values with the percentage of perovskites,red represents all B/B′ sites are d0 or d10 metal ions,grey represents only one of B/B′ sites is d0 or d10 metal ion,blue represents none of B/B′ sites are d0 or d10 metal ion;(b) the perovskite bandgap distribution diagram,colored area represents visible light energy range;(c) pie chart of the distribution ratio of different B/B′ site ions;(d) comparison of bandgap values of 3 different structures,B/B′ sites are all with d0 or d10 metal ions.
从上述统计结果可以看出,对于适合光催化材料的寻找可以从B/B′为d10的钙钛矿入手,还可以通过替换B/B′位d10离子为d0离子来进一步扩大带隙值选择范围,同时钙钛矿结构对称性也可以用来进一步调节带隙的大小,这为实验上寻找新型全解水光催化材料提供了理论指导.
3.4 DFT 验证和全解水光催化剂预测
在讨论元素组成与构型对于双钙钛矿带隙的影响后,还需要对预测的结果进行进一步验证,并通过计算电子能带结构,获得带边位置满足光催化全解水的双钙钛矿材料.选用预测集中B/B′位为d0/d10金属离子且结构稳定(容忍因子t> 0.85)的菱方类结构材料进行DFT 验证.对结构保持稳定未发生畸变的29 种材料计算了能带结构,DFT所得带隙与机器学习预测带隙符合得较好,R2> 0.8(见图5(a)).
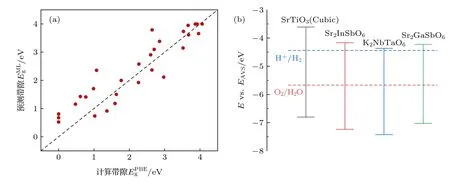
图5 (a) 29 种菱方类结构双钙钛矿材料DFT 带隙与机器学习预测带隙的比较;(b) 3 种候选双钙钛矿相对于水的氧化还原势的HSE 带边位置,以及作为比较基准的SrTiO3 (立方相)带边位置Fig.5.(a) DFT bandgap verification of 29 rhombohedral double perovskites in the prediction set;(b) the HSE band edge positions with respect to the water reduction and oxidation potential levels of selected double perovskites.SrTiO3 (cubic) is listed as a benchmark.
对上述29 种材料PBE 带隙分析后,选出带隙值处在0.8—1.8 eV 之间,且不含铅、汞离子的Sr2GaSbO6,Sr2InSbO6和K2NbTaO6这3 种双钙钛矿结构进行HSE 能带的计算.从表1 中可以看出,这3 种双钙钛矿候选材料的HSE 带隙都在可见光范围内,且HSE 带隙比其PBE 带隙值要大40%—50%.其中Sr2GaSbO6和Sr2InSbO6为间接带隙半导体材料,而K2NbTaO6为直接带隙半导体材料.一般来说,具有直接带隙的催化剂通常具有较高的电子空穴传输能力,它们能更有效地进行光子收集[49].因此,从带隙类型来看K2NbTaO6双钙钛矿材料相较于另外两种材料更适合作为光催化剂.另一方面,要实现可见光范围内的光催化全解水,除了其带隙值需要在可见光能量范围内,半导体材料的价带顶还要低于氧化电势(VH2O/O2=—5.67 eV),导带底也要高于还原电势(VH2/H+=—4.44 eV).参照立方相SrTiO3的能带结构,将3 种双钙钛矿O 的深能级与前者的对齐[50-52],可以得到K2NbTaO6,Sr2InSbO6和Sr2GaSbO6相对于水的氧化还原势的带边位置,如图5(b)所示.此3 种双钙钛矿氧化物带隙值在3 eV 左右,对应可见光吸收范围虽然较小,但这3 种材料的带边位置均满足光催化全解水的带边位置要求,合成材料价格低廉,不含重金属元素.因此,综合考虑了以上几点,它们很有可能是好的光催化剂材料.
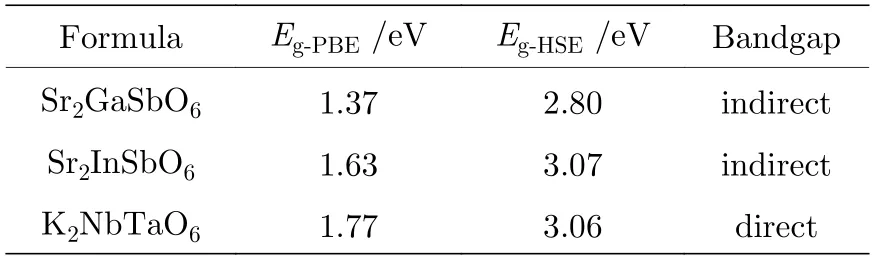
表1 3 种候选 双钙钛 矿材料 的PBE 带 隙、HSE 带隙及带隙类型Table 1. PBE and HSE bandgap of three kinds of double perovskite candidates and their bandgap categories.
4 总结
本文通过结合机器学习方法与第一性原理计算,设计了一套快速筛选功能材料框架,成功从56894 种候选材料中筛选出7909 种带隙符合光催化全解水需求的双钙钛矿氧化物材料.机器学习分类和回归的模型的准确率均接近90%.此外,对预测结果的统计分析表明,从B/B′位选择含有d10金属离子的双钙钛矿材料,更有可能找到符合光催化全解水带隙要求的材料,同时,将B/B′位的d10金属离子替换为d0金属离子可以调节带隙大小.最后,通过比对HSE 带边位置和全解水所需的氧化还原电势位置,挑选出Sr2GaSbO6,Sr2InSbO6和K2NbTaO6三种适合作为全解水光催化剂的双钙钛矿氧化物候选材料.本工作为快速、准确筛选设计双钙钛矿光催化剂提供了新的思路.
感谢东南大学大数据中心和天津国家超级计算中心的计算资源.
附 录
