“酶+X”耦合催化CO2资源化转化
2022-09-13刘书松张博禹吴振华赵熙瑶石家福
刘书松,张博禹,吴振华,赵熙瑶,石家福
(天津大学 环境科学与工程学院,天津 300072)
0 引 言
CO2是迄今为止排放量最多的温室气体,也是气候变化的主要驱动力[1-2]。由于CO2排放量增加而导致的全球变暖已成为人类面临的主要问题。据统计,全球化石燃料燃烧产生的CO2排放量在过去50多年增加了1倍多,每年超过30 Gt的CO2排放量导致全球变暖日益严峻[3]。国际能源情报署(EIA)预测,如果继续使用化石燃料作为主要能源供应来源,2030年CO2排放量将增至40.2 Gt[4]。研究表明,极端天气频发与大气中CO2浓度不断上升密切相关[5-8]。除全球变暖的净影响外,CO2还是导致海洋酸化的罪魁祸首[9]。大量CO2溶于海水生成碳酸(H2CO3)导致海水pH降低,进而破坏海洋的微碱性环境(pH约8.1),严重损害海洋生物多样性与生物地球化学循环过程[10]。
CO2排放量不断增加已成为影响全球的普遍性危害,为了保护环境免受全球变暖的影响,迫切需要国际间合作行动以限制气候变化。2020年,196个缔约国提交了国家自主贡献和实施战略,以实现《巴黎协定》确定的目标。2020年9月,我国在第75届联合国大会上提出了中国作为负责任大国应对全球气候变化的“30·60”目标,做出了2030年实现“碳达峰”,2060年实现“碳中和”的承诺。2021年11月,在第26届联合国气候变化会上50个国家承诺发展低碳卫生系统,14个国家设定了在2050年或之前实现净零碳排放的目标。国际社会法律基础的形成,加速了CCS发展步伐。CCS旨在将CO2从大气中隔离,并以一种永远不会排放的方式储存起来,如地质封存、海洋储存等[11-12]。然而,CCS仅能实现CO2在物理空间上的转移,经济负担较重[11]。此外,每年捕集的CO2数量巨大,需大量储存空间,存在泄漏风险[13-14]。YANG等[15]报道了一种基于超强碱/聚乙二醇进行CO2捕集—活化—转化的策略,将CO2从废物转化为增值化学品,为CO2治理提供了新范例。由于可通过生产附加值产品来提高CO2捕集过程的经济性,CCUS被认为是CCS的有效替代和补充方案[16]。将CO2转化为具有环境、经济和社会效益的产品以实现资源化利用,有望使CO2成为循环经济的新焦点并刺激新技术、新产品和新产业的发展。

笔者综述了近年来“酶+X”耦合催化CO2资源化转化相关研究工作,包括“酶+酶”耦合催化CO2资源化转化、“酶+化学”耦合催化CO2资源化转化、“酶+光”耦合催化CO2资源化转化和“酶+电”耦合催化CO2资源化转化,并对其系统特点进行分析。在结构解析基础上,讨论了“酶+X”耦合催化CO2资源化转化系统的关键设计原则,最后对未来研究需求进行展望和建议。
1 “酶+X”耦合催化CO2资源化转化
1.1 “酶+酶”耦合催化CO2资源化转化
“酶+酶”耦合催化CO2资源化转化又称为多酶催化CO2资源化转化,催化过程涉及2个或多个酶参与,第1种酶催化CO2转化形成的产物(中间体、中间产物)作为后续酶的底物或底物之一继续参与反应。多个酶的参与丰富了CO2转化为目标产物的路径可设计性,可显著缩短反应步骤、抑制中间产物积累和提高整体反应效率。与单酶催化系统相比,“酶+酶”耦合系统可以通过多步反应促使CO2还原产物向具有更高附加值的方向转化,极大提升CO2资源化转化的经济效益。1999年,OBERT等[33]首次报道了基于3种不同脱氢酶(酸脱氢酶、醛脱氢酶、醇脱氢酶)连续酶促还原CO2为甲醇的“酶+酶”耦合催化路径。该路径首先由甲酸脱氢酶(FateDH)催化CO2还原为甲酸盐,然后由甲醛脱氢酶(FaldDH)将甲酸盐还原为甲醛,最后通过乙醇脱氢酶(ADH)将甲醛还原为甲醇。该路径被认为是最经典的多酶催化CO2资源化转化的案例之一。为保持酶催化剂活性、协调多个酶催化过程间物质传递行为以及增强系统稳定性,“酶+酶”耦合催化系统常需对酶进行固定。如笔者课题组通过Pickering乳液和溶胶-凝胶法的协同作用,构建了一种基于纳米颗粒稳定微囊(NPSCs)的分隔式“酶+酶”耦合系统催化CO2转化至甲醛[34](图1)。该系统中,甲酸脱氢酶(FateDH,第1种酶)和甲醛脱氢酶(FaldDH,第2种酶)分别通过物理包埋在微囊壁内和化学耦合在微囊壁表面进行固定。坚固的微囊壁使该“酶+酶”耦合催化系统具有合适的微环境和卓越的稳定性。反应平衡后,固定化体系的甲醛产率为50%~60%,是同等反应条件下游离体系的1.47~1.76倍。此外,该系统循环使用10次后甲醛产率基本保持不变。此外,还通过仿生矿化和仿生黏合法构建了一种基于超薄明胶-二氧化硅杂化微囊(GelCSi)固定化的三酶耦合系统催化CO2转化至甲醇[35](图2)。在该系统中甲酸脱氢酶(FateDH,第1种酶)、甲醛脱氢酶(FaldDH,第2种酶)和乙醇脱氢酶(ADH,第3种酶)分别通过物理包埋在微囊内腔、共价连接到儿茶酚修饰的明胶层和物理包埋在硅胶层被固定。GelCSi微囊能在纳米距离内精确放置酶级联的底物/中间体,并根据每种酶的活性合理调节酶的数量。微囊的超薄壳层和介孔结构(厚度70 nm,孔径3.6 nm)加速了CO2与中间产物分子的传输过程。反应达到平衡时甲醇产率和选择性分别为71.6%和86.7%,显著高于游离酶体系(35.5%、47.3%)。同时,GelCSi微囊较高的机械稳定性为该耦合催化系统提供了良好的可回收性和适宜储存的微环境。
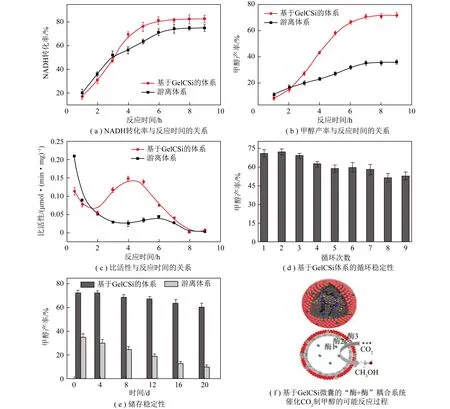
图2 基于GelCSi固定化的三酶耦合催化系统[35]Fig.2 Three-enzyme coupled catalytic system based on GelCSi immobilization[35]
多个酶的耦合为CO2资源化转化路径设计提供了可能,进一步丰富了目标产物种类的多样性。TONG等[36]开发了一种利用CO2和乙醇合成L-乳酸的“酶+酶”耦合催化体系,并巧妙地嵌入了一个内部辅因子再生循环,消除了辅因子再生所需的额外化学物质或能量(图3,vi为可逆反应的反应速度,-为反应的逆向)。在间歇反应中通过保持乙醇浓度恒定驱动L-乳酸生成,实现了4 d内乙醇添加量转化率为41%,乳酸物质的量浓度达到87 μmol/L。该合成路线为CO2资源化转化提供了一条新的反应途径。SCHWANDER等[37]利用来自9种不同生物体的17种酶建立了一个在体外连续转化CO2的合成循环——巴豆酰辅酶A(辅酶A,CoA)/乙基丙二酰辅酶A/羟丁酰辅酶A(CETCH)循环(图4,M代表未标记,M+x代表检测到x处标记,虚线框中均为酶)。CETCH循环能以速率5 nmol/(mg·min)(以蛋白质计)将CO2转化为有机分子,比自然生物系统效率高20%。CETCH循环在6个自然进化的CO2固定途径的基础上增加了第7个人工合成的替代途径,从而为“酶+酶”耦合催化CO2资源化转化开辟了新道路。

图3 CO2合成L-乳酸的酶促反应路线[36]Fig.3 Enzymatic reaction route for synthesis of L-lactic acid from carbon dioxide[36]
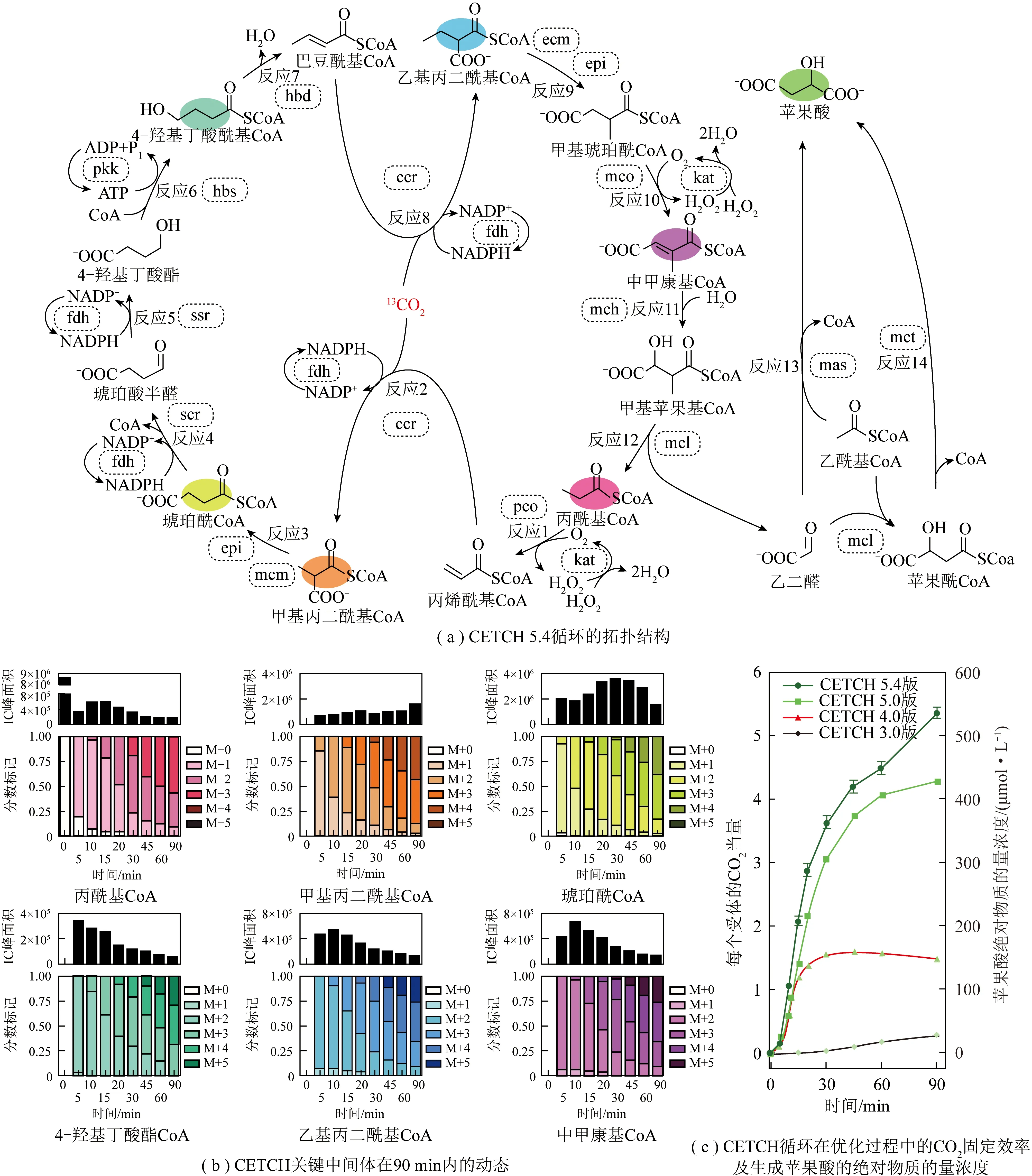
图4 CETCH循环[37]Fig.4 CETCH cycle[37]
1.2 “酶+化学”耦合催化CO2资源化转化
“酶+化学”耦合催化CO2资源化转化涉及化学催化和酶催化2个催化过程:① 在化学催化过程中,CO2作为原料经化学催化剂催化转化为C1中间体(如HCOOH、CH3OH和CH3CHO等);② 甲酸等C1中间体作为底物或底物之一参与酶催化过程,最终转化为目标产物。整体反应过程无需引入光或电等外部能量,具有能耗低、反应路径可设计、产物立体选择性高等特点。得益于化学催化过程对CO2的预先转化,酶催化过程可直接以C1化合物作为反应起点。因此“酶+化学”耦合系统在催化CO2转化为C2/C2+等多碳化合物方面表现出独特优势。CAI等[38]报道了首条人工淀粉合成代谢途径(ASAP),该系统耦合化学催化与多酶催化过程构成了11个核心反应:CO2首先在氢气驱动下经化学催化剂ZnO-ZrO2催化转化为甲醇,之后甲醇通过改造的10种核心多酶组成的级联催化路径转化为淀粉(图5、6,agp-M1、agp-M2、agp-M3为agp的3种变体,fbp-AR为fbp的变体,fbp-AGR为fbp-AR与抗G-6-P变体组成的复合变体)。ASAP资源化转化CO2速率高达22 nmol/(mg·min)(以催化剂计),较自然界淀粉合成速率提升了750%。DESMONS等[39]报道了一种新颖且以CO2为原料合成二羟基丙酮或手性化合物L-赤藓酮糖的“酶+化学”耦合催化路径(图7)。CO2首先通过铁催化的硼氢化反应选择性还原为双(硼基)缩醛化合物,该化合物在30 ℃水溶液中易水解生成甲醛。随后甲醛中间体参与选择性酶转化,在甲醛酶催化下生成二羟基丙酮,或通过甲醛酶和6-磷酸果糖缩醛酶的级联反应转化为具有高立体选择性的L-赤藓酮糖。酶催化过程和化学催化过程耦合,为实现CO2高值转化和从头合成多碳生物质开辟了新道路。
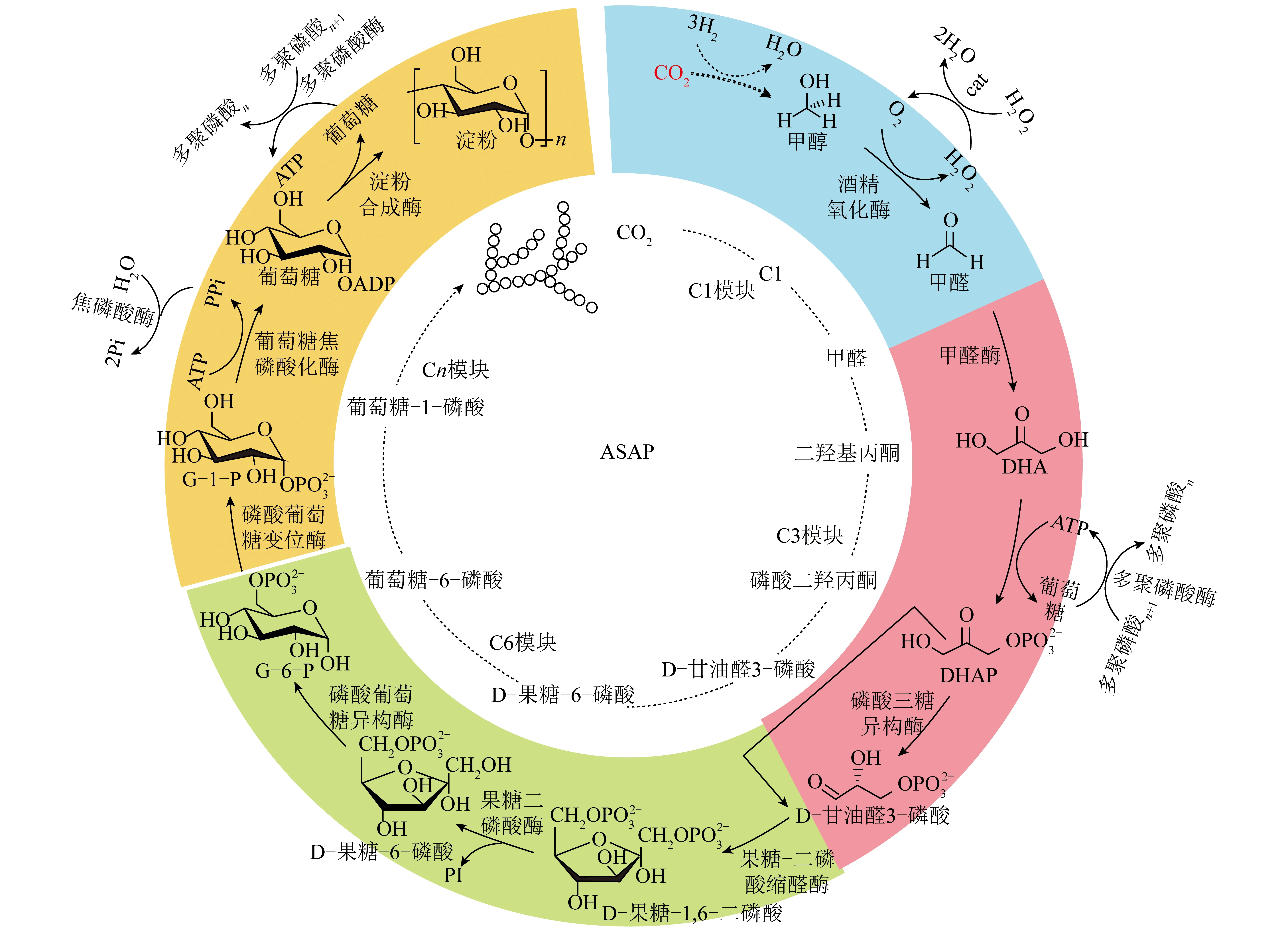
图5 人工淀粉合成途径的设计和模块组装[38]Fig.5 Design and modular assembly of an artificial starch anabolic pathway[38]
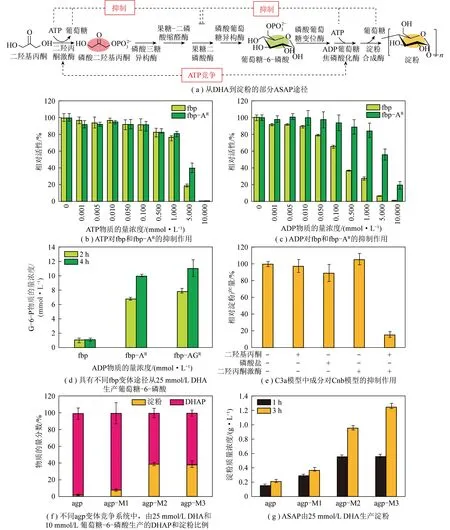
图6 解决ASAP中的主要瓶颈[38]Fig.6 Resolving main bottlenecks in ASAP[38]
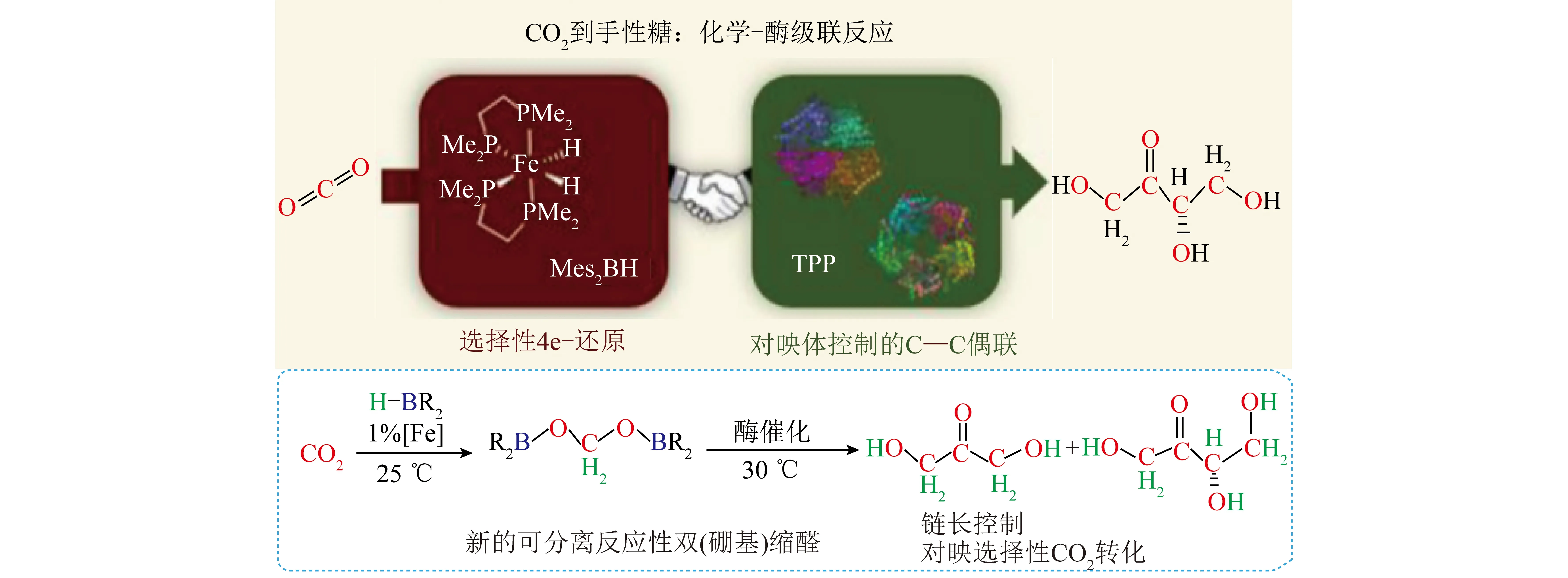
图7 “酶+化学”耦合催化CO2对映选择性还原为L-赤藓酮糖[39]Fig.7 Enantioselective reduction of CO2 to L-erythrulose catalyzed by "Enzyme + Chemo" coupling system[39]
1.3 “酶+光”耦合催化CO2资源化转化
“酶+光”耦合催化CO2资源化转化是模拟自然界绿色植物光合作用,以CO2为原料、以半导体和酶为催化剂、以辅酶等氧化还原当量为能量载体,通过耦合光催化辅酶再生反应和酶催化CO2转化反应实现CO2高值转化的绿色生物制造过程。具有反应条件温和可控、光-酶模块间物质/能量传递机制易解析、路径可放大等特点。该系统由于可实现辅酶的循环再生,避免了辅酶依赖型酶催化反应对化学计量的外源性还原当量的消耗,大幅降低了反应成本。此外,能量外场(光能)的引入促使CO2转化产物多为酶促加氢后的载能化合物(如甲醇等)。然而,光催化剂、酶催化剂和辅酶的时空共存增加了系统的复杂性。协调优化光催化剂和酶催化剂的相容性和光催化反应-酶催化反应间的能量传递行为是实现CO2高效资源化转化的关键[40-41]。
通过隔室设计将光催化剂和酶催化剂在物理空间上限域分隔是提高相容性的有效手段。受自然界类囊体结构启发,笔者课题组基于鱼精蛋白-二氧化钛(PTi)微囊作为限域载体构建了一种“人工类囊体(CdS/PTi)”[42](图8,h为普朗克常数,ν为频率,TEOA为三乙醇胺)。尺寸选择性的微囊成功将CdS量子点和甲酸脱氢酶(CbFDH)分别限制在囊腔内和囊壁外,避免了CbFDH和CdS量子点的直接接触,CbFDH的相对活性在光照1 h后保持(96.1±5.3)%。作为对比,在PTi微囊壁外表面沉积CdS量子点制备了PTi/CdS微囊。对于PTi/CdS微囊,CdS量子点可直接接触CbFDH,致使CbFDH的相对活性在光照1 h后急剧降至(71.9±1.4)%。表明隔室设计能提高光催化剂与酶催化剂的相容性。得益于“人工类囊体”的合理结构设计,该“酶+光”耦合体系在单酶(CbFDH)和多酶(TsFDH-FaldDH-YADH)作用下,CO2转化为甲酸盐和甲醇的初始生产速率分别为1 500和99 μmol/(L·h)。
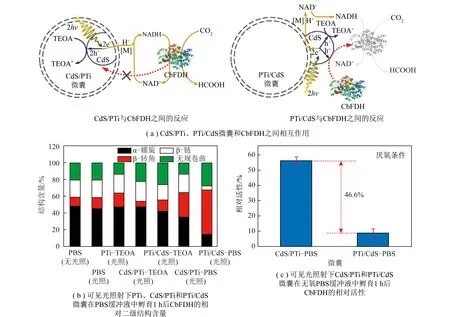
图8 “人工类囊体”协调光催化剂和酶催化剂相容性[42]Fig.8 "Artificial thylakoid" coordination the compatibility of photocatalysts and enzymes[42]
光催化反应-酶催化反应间的能量传递主要表现为辅酶等氧化还原当量的光催化再生和酶催化消耗循环。这个过程的关键在于协调强化光生电子的产生—传递—利用3个步骤。为强化光生电子的产生,一般选择具有较宽吸收光谱的半导体作为光催化剂,并进行一定修饰提高光捕获能力。ZHANG等[43]利用具可见光响应的石墨相氮化碳(g-C3N4)作为光催化剂,通过焙烧、剥离和共价键构建等方法在其表面修饰均苯四甲酸二亚胺(PDI)(图9,k为斜率,C0为NAD+初始浓度,C为NADH浓度)。PDI有机分子有效拓宽了原始g-C3N4的可见光响应范围,光吸收边缘拓宽到约660 nm,辅酶再生转化频率达0.242 h-1。PDI/g-C3N4与甲酸脱氢酶耦合实现了CO2到甲酸的定向转化,反应9 h后甲酸收率达114.8 μmol/L,生产效率达1.269 mmol/(g·h)。光生电子传递过程的强化主要依赖于电子传递链的设计或额外施加偏压(光电化学电池,PEC)。JI等[44]将同轴电纺/电喷雾和逐层自组装相结合,构建了基于中空纳米纤维的“酶+光”耦合CO2转化系统(图10)。该系统中光敏剂、电子介体和3种脱氢酶(甲酸脱氢酶、甲醛脱氢酶和乙醇脱氢酶)在纳米尺度上精确排列,确保了光生电子在各个活性组分间有效传输。与基于溶液的系统相比,该高度集成系统的CO2转化率从35.6%提高至90.6%。光照反应10 h后甲醇产量达60.39 μmol/L。KUK等[45]利用赤铁矿光阳极(a-Fe2O3)和铋铁氧体光阴极(BiFeO3)制备了一种用于可见光辅助再生烟酰胺辅因子(NADH)的氧化铁基串联PEC电池(图11)。该PEC电池通过在光阴极集成一个由3种脱氢酶构成的级联系统(甲酸脱氢酶-甲醛脱氢酶-乙醇脱氢酶)实现了“酶+光”耦合催化CO2转化为甲醇。得益于外部偏压作用下光生电子的快速定向传递,甲醇生产效率明显高于其他可见光系统。施加0.8 V外部偏压的条件下,6 h内甲醇产量达到约1.32 mmol/L。光生电子利用过程的强化可以通过缩短电子传递距离来实现。受自然界铁氧还蛋白-NADP+氧化还原酶(FNR)固载于类囊体膜上的启发,笔者课题组通过多酚诱导的仿生黏合法,在氮化碳(PCN)表面生长一层固载有辅酶再生反应催化中心([Cp*Rh(bpy)H2O]2+,Rh)的纳米壳层(TA/PEI-Rh),制备了PCN@TA/PEI-Rh核壳光催化剂[46](图12)。辅酶再生反应催化中心Rh在PCN表面的固载缩短了光生电子在Rh和PCN间的传递距离,不仅强化了光催化辅酶再生过程的电子利用部分,还简化了该辅酶再生反应体系。进一步将PCN@TA/PEI-Rh与甲酸脱氢酶(FDH)耦合,可以实现“酶+光”耦合催化CO2转化制甲酸的过程,光照2 h后可产生甲酸2.1 mmol/L,选择性为100%。能量传递过程中光生电子的产生—传递—利用是一个连续反应,系统构建中一般会同时强化其中2个或3个步骤以实现CO2到有价值化学品的高效转化。
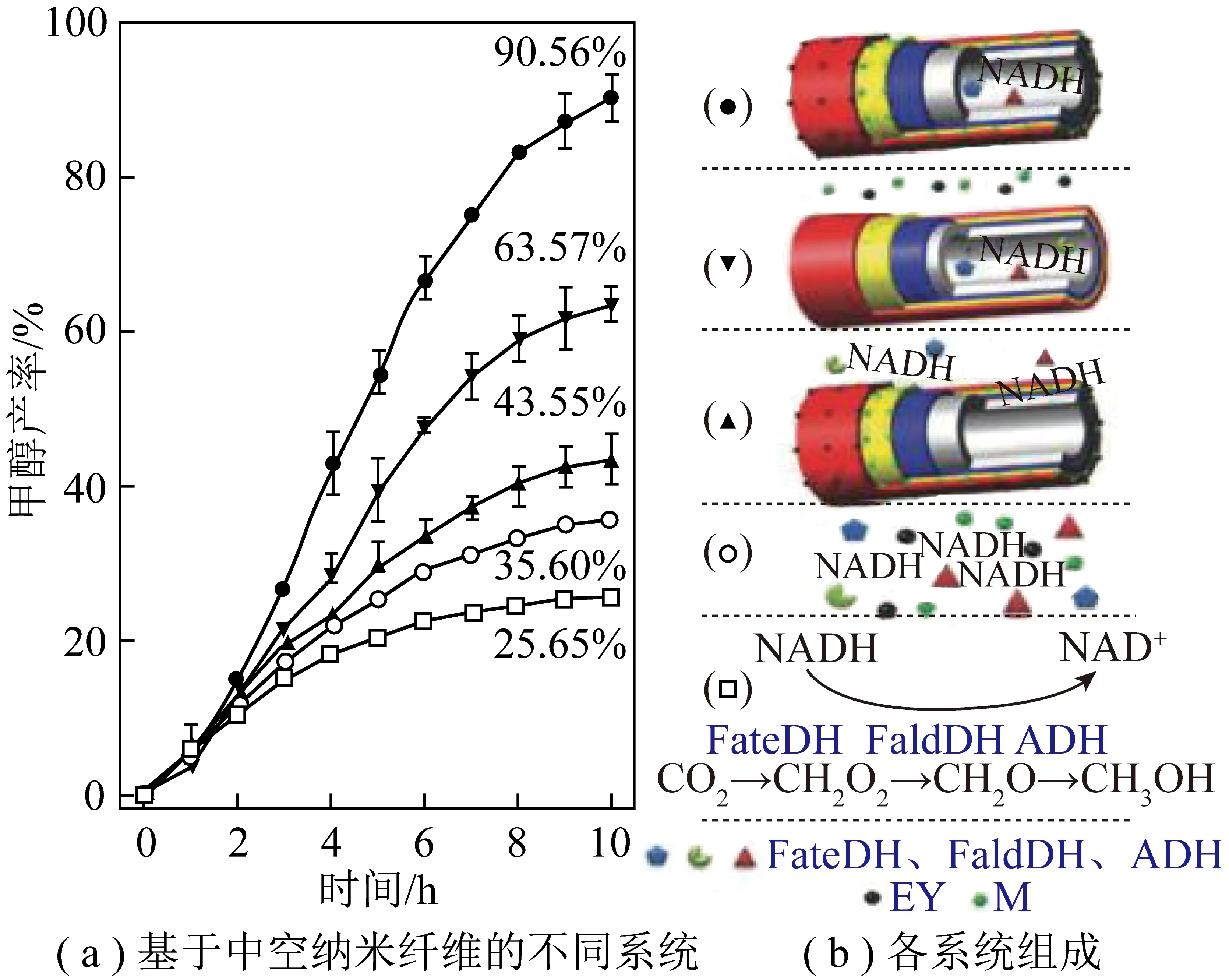
图10 基于聚电解质掺杂中空纳米纤维的“酶+光”耦合催化系统用于CO2合成甲醇[44]Fig.10 "Enzyme + Photo" coupled catalytic system based on polyelectrolyte-doped hollow nanofibers for methanol synthesis from CO2[44]
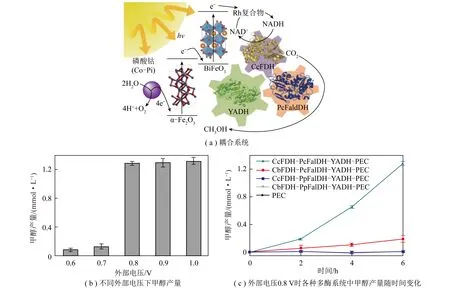
图11 光能驱动CO2和水通过3种酶(CcFDH、PcFaldDH、YADH)级联生产甲醇[45]Fig.11 Solar-assisted production of methanol from CO2 and water through a three-enzyme (CcFDH,PcFaldDH,YADH) cascade[45]
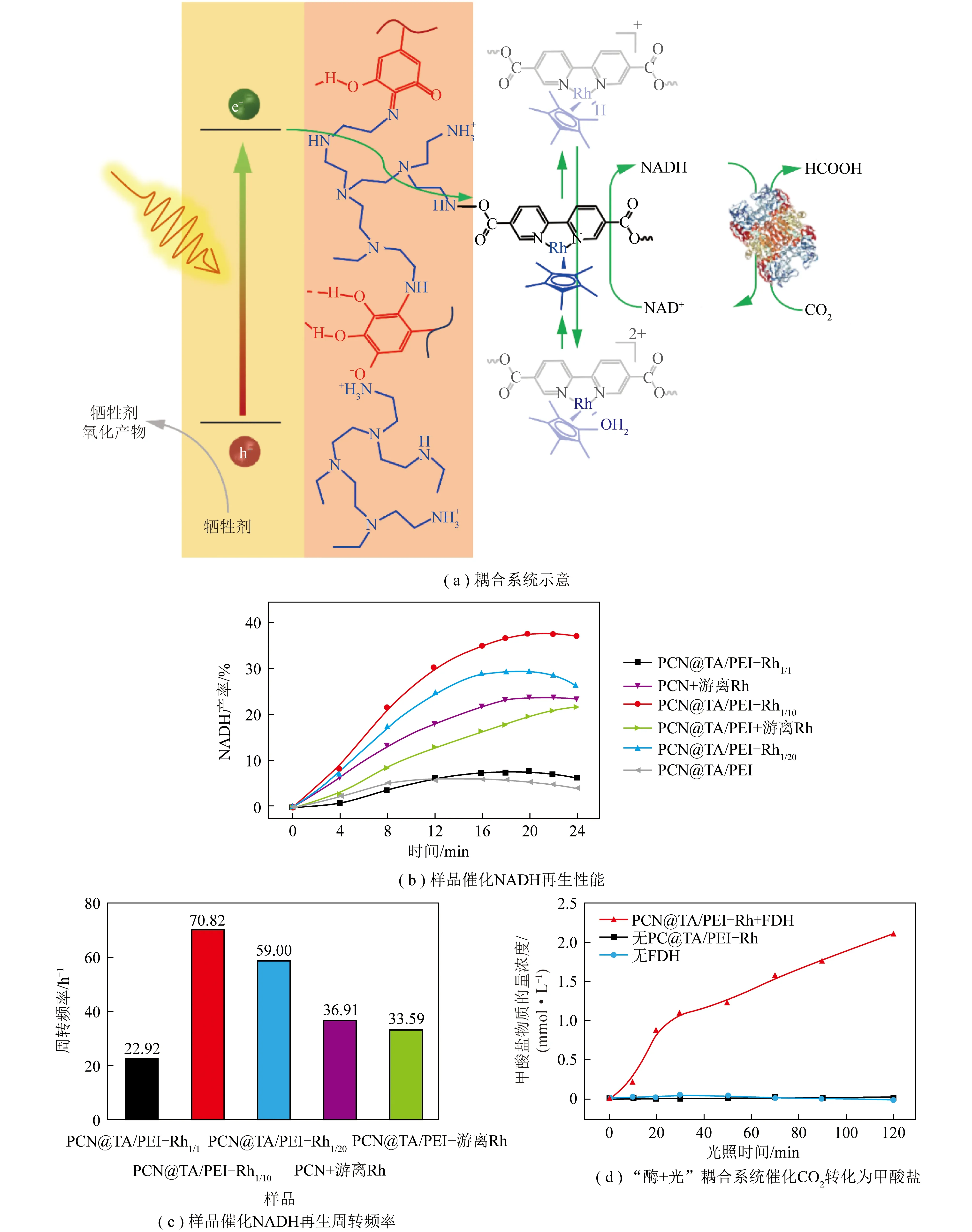
图12 PCN@TA/PEI-Rh耦合甲酸脱氢酶(FDH)催化CO2制甲酸[46]Fig.12 PCN@TA/PEI-Rh coupled FDH catalyzing CO2 to formate[46]
1.4 “酶+电”耦合催化CO2资源化转化
“酶+电”耦合催化CO2资源化转化在系统组成上与“酶+光”耦合系统的PEC类似,但该系统对电极材料的光响应特性并不作要求。在外部电压的作用下,阳极通过电解水反应为阴极提供电子和质子用于间接(NADH等辅酶再生)或直接驱动酶促CO2转化。相较于“酶+光”耦合系统利用辅酶作为电子载体,“酶+电”耦合系统可通过施加外部偏压直接实现电极与酶活性中心之间的有效电子迁移。能量外场(电能)的引入使得该系统同样广泛应用于CO2酶促加氢合成载能化合物。为了提高系统的整体催化性能,需对阴极酶促模块进行精密设计。SCHLAGER等[47]采用碳毡为阴极电极材料,并利用海藻酸盐-硅酸盐杂化凝胶基质将甲酸脱氢酶、甲醛脱氢酶和乙醇脱氢酶共固定在该电极上开发了一种无需辅酶的“酶+电”耦合还原CO2合成甲醇的催化系统(图13)。通过将电子直接从电极注入到共固定化的3种脱氢酶来取代昂贵的辅酶NADH,实现了CO2到甲醇的直接还原。在恒定电位为±1.2 V时,检测到约0.15×10-6(0.005 mmol/L)甲醇,法拉第效率(Faraday efficiency,FE)约为10%。ZHANG等[48]通过将ZIF-8固载的级联多酶模块、NADH再生模块与Rh复合物接枝电极相结合,开发了一种有效的“酶+电”耦合还原CO2合成甲醇的催化系统(图14)。该系统中,Rh复合物接枝电极对NAD+的还原表现出良好的稳定性和高效的电子转移,NAD+电催化生成NADH的选择性接近100%。ZIF-8固载的级联多酶模块不仅提升了酶的稳定性,还有效浓缩了底物(CO2)和NADH,提高了整体催化效率。在0.7 V电压作用下,反应3 h后甲醇物质的量浓度达到0.742 mmol/L,生产速率达822 μmol/(g·h)。

图13 “酶+电”耦合催化系统无辅酶催化CO2转化为甲醇[47]Fig.13 "Enzyme + Electro" coupled catalytic system catalyzes CO2 to methanol without coenzyme[47]
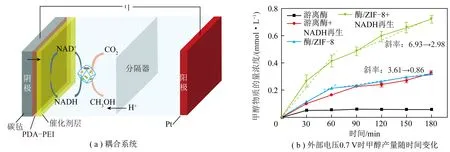
图14 “酶+电”耦合催化CO2转化为甲醇[48]Fig.14 "Enzyme + Electro" coupling catalyzes CO2 to methanol[48]

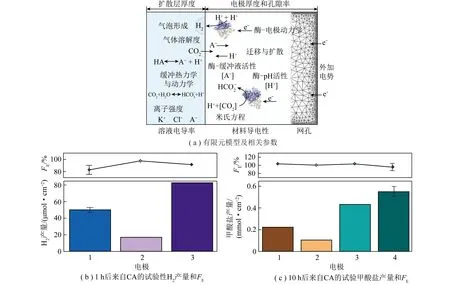
图15 “酶+电”耦合催化系统中酶在受限电极空间的局部微环境调控[56]Fig.15 Local microenvironment regulation of enzymes in confined electrode spaces in the "Enzyme + Electro" coupled catalytic system[56]
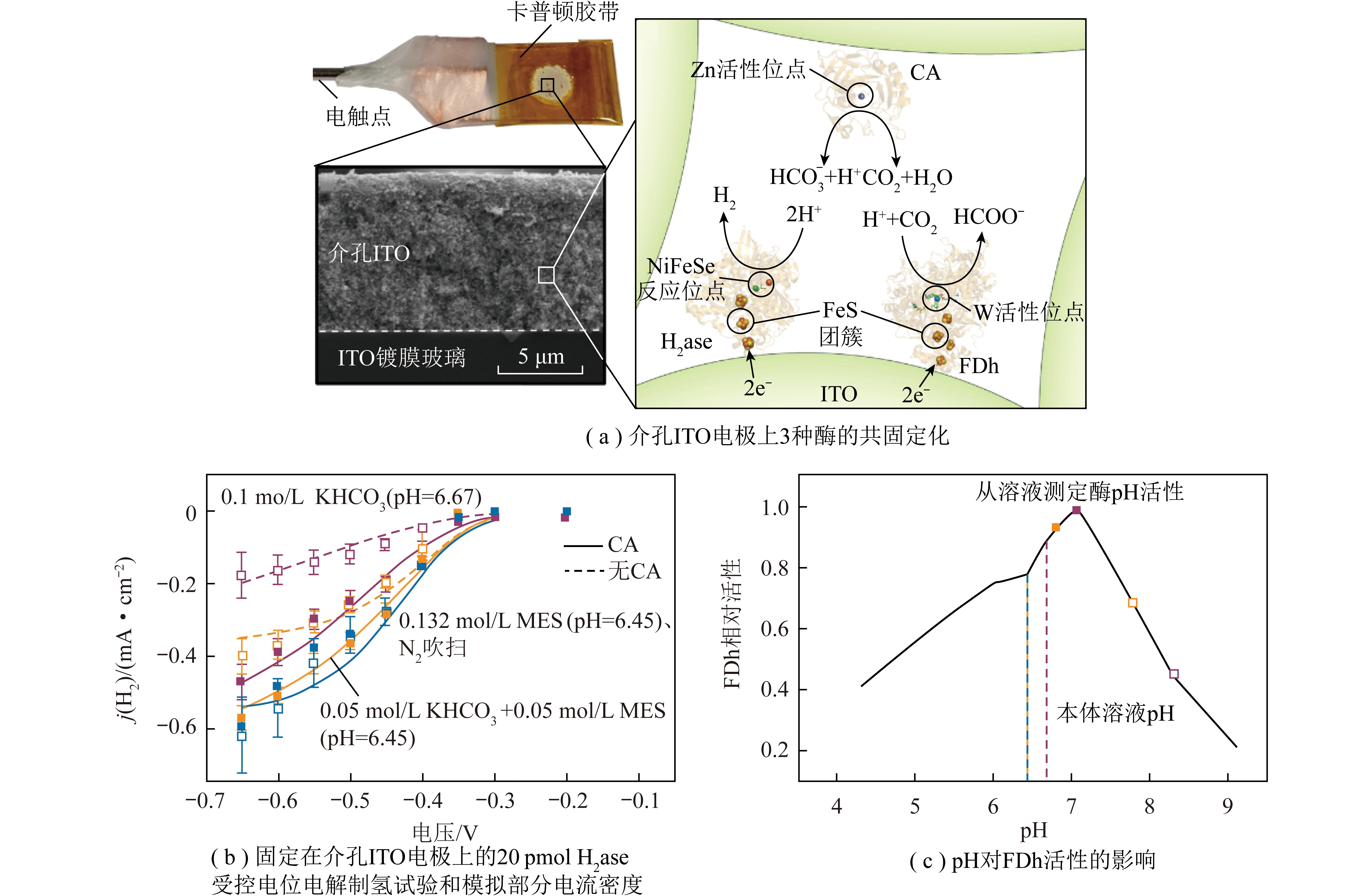
图16 CO2水合对“酶+电”耦合催化系统局部微环境的影响[57]Fig.16 Effects of CO2 hydration on the local microenvironment of the "Enzyme + Electro" coupled catalytic system[57]
2 结语与展望
将CO2转化为高值化学品或燃料是提高碳捕集过程经济性、实现“碳中和”的关键。基于酶催化反应的耦合催化系统为CO2资源化转化创造了丰富、可设计的路径网络,在扩大CO2利用市场规模方面表现出巨大的应用潜力。发展“酶+X”耦合催化CO2资源化转化技术不仅能遏制温室效应,更能将CO2由废物转化为宝物,形成一种可循环的CO2管理模式。然而,“酶+X”耦合催化系统同样存在不足:
1)生物催化剂驱动力不足致使酶催化反应过程成为系统的主要限速步骤,进而导致CO2整体资源化转化效率较低。
2)酶催化过程与其他催化过程在反应条件上存在巨大差异,耦合系统各模块间物质/能量传递受阻,兼容性较差。仅依赖于“酶+X”耦合催化对环境中CO2进行大规模资源化转化基本不现实,该技术更多作为CCS的有效替代和补充方案,地质/海洋封存仍是必要选择。
为进一步扩大CO2利用市场规模,“酶+X”耦合催化CO2资源化转化技术必须在现有基础上实现突破:
1)需发展新技术保持酶的活性。酶作为一类生物催化剂,其化学本质是蛋白质或核酸,极易受反应环境(包括温度、酸碱度、自由基等)的影响而失活。开发新的酶保护技术对保持酶活性、提高耦合系统稳定性等具有重要意义。
2)需发展关键酶元件挖掘与定向改造技术。酶的催化反应速率在一定程度上限制了耦合系统的整体CO2还原效率。利用基因组学和大数据分析挖掘关键酶基因序列并开展基于机器学习、计算机虚拟突变的酶理性改造研究,研发具有优异工业属性的酶突变体,是提升整体CO2资源化转化效率的有效方法。
3)需在现有系统上构建碳富集模块。当前“酶+X”耦合系统催化的底物碳一般为经捕集、浓缩后的高纯CO2,而环境中的CO2具有高分散、不均一、低浓度等特点。在现有系统的基础上构建碳富集模块,直接捕集大气中的CO2并进行资源化转化十分必要。
4)需对系统进行工程放大。目前已报道的“酶+X”耦合催化CO2资源化转化系统多局限于实验室规模的科学探讨,仅有极少量路径实现工业化。将现有系统放大,走出实验室并真正应用到CCUS,仍是未来研究工作的重点。
