伤寒沙门菌非编码RNA617的分子鉴定及其对生物膜形成的影响
2022-08-30靳梦彤李雪党娟娟李泽宇张盈黄新祥
靳梦彤,李雪,党娟娟,李泽宇,张盈,黄新祥
1. 江苏大学医学院生物化学与分子生物学系,江苏 镇江 212013; 2. 南通市第三人民医院检验科,江苏 南通 226006
伤寒沙门菌(Salmonellaentericaserovar Typhi,S. Typhi)是沙门菌属中的一种重要的肠道致病菌,人是其天然宿主[1]。由伤寒沙门菌引起的伤寒热,主要迹象为发热、血培养或肥达反应阳性等。伤寒沙门菌可通过污染的食品或水经口感染,抵御宿主免疫屏障在其体内大量繁殖,且随血流播散至实质器官中,引发多种临床症状[2]。S. Typhi 形成的细菌聚集膜样物,即生物膜,可抵御宿主免疫的杀伤和外界环境的刺激,从而利于自身传播及在体内定植[3-4]。生物膜的主要成分包括细菌本身和其分泌的多糖基质和多种蛋白,其形成过程受复杂的调控网络控制,包括双组份调节系统、全局调节因子、非编码RNA(non-coding RNA,ncRNA)[5-6]等。ncRNA指不编码蛋白质的RNA,一般通过与靶RNA互补配对或者改变靶标构象的方式发挥调节作用[7-8]。
本课题组前期通过RNA-Seq技术对生物膜状态和浮游状态的S. Typhi进行转录组分析,发现多个在2种状态下差异表达的ncRNA[9],其中ncRNA617在生物膜细菌中的表达丰度明显高于浮游菌。鉴于此,本研究对ncRNA617进行分子鉴定,以探究其在生物膜形成过程中发挥的作用, 为后续机制研究提供基础。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒伤寒沙门菌野生菌株S. Typhi GIFU10007、大肠埃希菌(Escherichiacoli,E.coli)λ372、DH5α和自杀质粒 pGMB151由日本岐阜大学医学院微生物学教研室馈赠,重组质粒pGMB151-F、ncRNA617过表达重组质粒、ncRNA617缺陷菌株(Δ617)、回补菌株(Δ617-c617)和过表达菌株(007-p617)、野生对照菌株(007-pBAD33)和缺陷对照菌株(Δ617-pBAD33)由本实验室制备。
1.1.2 试剂胰蛋白胨、酵母提取物、琼脂粉均购自英国OXOID公司,氨苄西林购自上海生工生物工程有限公司,TRIZOL、氯仿均购自美国Sigma公司,限制性核酸内切酶Bam H I、T4 DNA连接酶、SMART RACE 5’/3’ Kit均购自宝生物工程(大连)有限公司,Phanta Super-Fidelity DNA聚合酶、逆转录试剂、SYBR荧光定量试剂盒均购自南京诺唯赞生物科技有限公司,96孔圆底细胞培养板购自美国Corning公司,DIG Northern Starter Kit购自瑞士Roche生物科技公司。
1.1.3 引物本研究所用引物信息如表1所示,引物由苏州泓讯生物科技有限公司合成。

表1 研究用引物Tab.1 List of primers

(续表1)
1.2 伤寒沙门菌ncRNA617的分子鉴定
1.2.1 RNA提取挑取细菌的单克隆在溶菌肉汤(lysogeny broth,LB)培养基中于37 ℃和250 r/min进行过夜培养,第2天将过夜培养物以1∶100比例稀释继续培养至吸光度A600值为0.4,4 ℃、4 000 r/min离心10 min后收集菌体,用TRIZOL法提取总RNA,具体方法参考文献[10]。
1.2.2 Northern blot实验依照已有方法[11]进行,依据ncRNA617表达高峰区域序列设计特异性探针引物制备探针,检查探针质量后保存待用。在6%的(尿素)变性聚丙烯酰胺凝胶上分离RNA(30 μg),经电转(300 mA,40 min)将凝胶上的RNA转移到硝酸纤维素膜上。用2×SSC(saline sodium citrate,SSC)缓冲溶液洗涤含有RNA的膜2次,洗去部分非特异性结合的探针,并在80 ℃下干燥2 h以固定RNA。随后,将膜用焦碳酸二乙酯(diethylpyrocarbonate,DEPC)水洗涤3遍,并将膜放入含ncRNA617特异性探针的杂交液中。杂交完成后经过洗膜、孵育地高辛抗体,再用化学发光成像系统进行曝光并拍照。
1.2.3 5’RACE使用SMART 5’RACE Kit进行cDNA 5’末端快速扩增反应,具体方法参考文献[12]。为获得第一链cDNA,在1 μg总RNA中加入基因特异性引物(gene-specific primers,GSP)和逆转录酶进行逆转录以获得cDNA,以通用短引物和基因特异性引物为引物,以cDNA为模板进行 5’RACE PCR反应,获取目的PCR产物。通过克隆将PCR产物插入基于pUC19载体改造的载体中。利用DNA测序确定ncRNA617的转录起始位点。
1.2.4 3’RT-PCR为探寻ncRNA617可能的转录终止位点,在ncRNA617表达高峰区域设计前向引物3’PA,在ncRNA617的下游设计后向引物 3’PB1~3’PB5。以各个后向引物为特异性引物对S. Typhi总RNA进行逆转录,再分别对前向引物进行PCR扩增,取适量产物进行电泳分析。
1.3 菌株构建
1.3.1 ncRNA617缺陷菌株构建依据文献[13]的方法构建缺陷菌株(Δ617),首先通过PCR从S. Typhi 野生菌株DNA中扩增出ncRNA617上下游同源臂片段F1和F2,用重叠PCR获得重组片段F。然后将含有限制性内切酶位点Bam H I的重组片段F克隆到自杀质粒pGMB151中。通过热激法将重组质粒pGMB151-F转入大肠埃希菌λ372感受态细胞中,涂布于含100 μg/mL氨苄青霉素的LB固体培养平板上,37 ℃培养12 h后挑取单菌落进行培养,提取质粒,经苏州泓讯生物科技有限公司测序验证,将阳性重组质粒在电压2.5 kV、电阻200~1 000 Ω条件下电转入S. Typhi野生菌株感受态,经5%蔗糖板传代筛选和普通LB平板稳定传代培养,最终获得缺陷菌株(Δ617)。
1.3.2 ncRNA617回补菌株和过表达菌株构建使用ncRNA617特异性引物PF/PR扩增目的片段,将含有限制性内切酶位点Xba I、Hind III的目的片段克隆至质粒pBAD33,将重组质粒热激转入大肠埃希菌DH5α感受态细胞中,涂布于含 34 μg/mL氯霉素的LB固体培养平板上,37 ℃培养12 h后挑取单菌落进行培养提取质粒,经双酶切和PCR验证,将疑似阳性质粒送至苏州泓讯生物科技有限公司进行测序验证,阳性重组质粒在电压2.5 kV、电阻200~1 000 Ω条件下电转入缺陷菌株和野生菌株,经质粒pBAD33验证引物YZF/YZR筛选,构建回补菌株(Δ617-c617)和过表达菌株(007-p617)。
1.3.3 野生对照菌株和ncRNA617缺陷对照菌株构建将pBAD33空质粒按照同样的电转条件电转入野生菌株和缺陷菌株,经质粒pBAD33验证引物YZF/YZR筛选,构建相应的野生对照菌株(007-pBAD33)和缺陷对照菌株(Δ617-pBAD33)。
1.4 伤寒沙门菌ncRNA617对生物膜形成的影响
1.4.1 生物膜形成测定使用胰酪胨大豆肉汤培养基(tryptic-soytone-broth-medium,TSB)培养相关菌株至A600值为0.4,取200 μL菌液,接种于96孔U型底细胞培养板,30 ℃静置培养96 h,进行生物膜形成量检测[10],培养结束后吸弃孔内菌液,用无菌磷酸缓冲液轻柔吹洗孔内3次,经甲醇固定后将培养板真空干燥20 min,加入 200 μL 结晶紫溶液后染色15 min,最后用蒸馏水清洗孔内3次,扣干水分后将板置于干燥箱内干燥20 min,加入 200 μL 30%乙酸溶液溶解孔内壁上的生物膜,采用酶标仪于A570波长测定对应孔内吸光度,测定值的高低代表细菌形成生物膜能力的大小。实验进行3次重复。
1.4.2 实时荧光定量PCR伤寒沙门菌生物膜的成分较多[14-15],包括鞭毛、菌毛、纤维素等。此次研究选定生物膜形成通路中与鞭毛、菌毛、胞外多糖、纤维素等相关的11个基因(见表2),采用qPCR分析相关基因的mRNA水平。TRIZOL法提取细菌总RNA,用4×gDNA Wiper消化除去基因组DNA,取2.5 μg RNA用随机引物进行逆转录得到cDNA,这些cDNA分别作为内参基因5s和生物膜形成相关基因的cDNA模板,进行qPCR实验,实验重复3次。

表2 生物膜形成相关基因Tab.2 Biofilm formation-related genes
1.4.3 生物信息学预测将ncRNA617和差异基因mRNA序列信息提交至IntaRNA系统,进行ncRNA617与差异基因的相互作用区域分析。
1.5 统计学方法
数据表示为平均值±标准偏差。用GraphPad Prism8对数据进行t检验分析,P<0.05代表有统计学意义。
2 结果
2.1 伤寒沙门菌ncRNA617的分子鉴定
在生物膜及浮游状态下,由S. Typhi野生株的转录组测序结果可知,在mig-14和t2681(假定基因)基因间区存在一段高峰表达区,该序列在生物膜S.Typhi 细菌中的表达丰度明显高于浮游S.Typhi 细菌。用对应特异性探针进行Northern blot检测,发现1条特异性杂交带,与RNA Marker对照得知其长度约300 nt(见图1A)。5’RACE PCR实验结果表明,泳道S有扩增出片段(见图1B),泳道U和G为阴性对照,均无条带,通过克隆将PCR产物插入pUC19载体改造的载体中,通过比对DNA测序与S.Typhi基因组序列信息(见图1C),结果提示该ncRNA转录起始位点位于mig-14终止密码子下游967 nt处。3’RT-PCR结果表明,片段长度为270 bp有阳性扩增,但长为452 bp、651 bp、887 bp、1123 bp的扩增为阴性(见图1D)。结合S.Typhi基因组的结构分析,提示该ncRNA转录终止位点位于t2681基因起始密码子上游 2 378~2 560 nt。综合Northern blot、5’RACE实验和3’RT-PCR实验的结果,可知ncRNA617长度在270~452 nt,从而绘制其基因结构位置图(见图1E)。

A. ncRNA617 was detected by Northern blot analysis using a specific DIG-labeled probe (M: Marker). B. 5’ RACE PCR product of ncRNA617 (S: sample; U: UPM only; G: GSP only;M: Marker). C. Blast analysis of the sequence. D. 3’ RT-PCR of the ncRNA617(1,4,7,10,13:Sample ;2,5,8,11,14:Blank control;3,6,9,12,15:Positive control; M: Marker). E. Genomic location and structure of ncRNA617.
2.2 ncRNA617缺陷菌株的构建
以S.Typhi野生株DNA为模板,分别进行PCR以获取目的片段上下游同源臂片段F1(272 bp)和F2(459 bp),如图2A所示,用重叠PCR获得重组片段F(731 bp)(见图2B),将重组片段F插入自杀质粒pGMB151的Bam H I位点,通过测序筛选阳性重组质粒,发现插入自杀质粒pGMB151的Bam H I位点的片段无突变缺失,并且与已知片段序列的信息一致性达100%(见图2C)。将阳性重组质粒pGMB151-F电转导入S.Typhi野生株,经5%蔗糖板传代培养筛选和普通LB平板稳定传代培养获得Δ617菌株(见图2D)。

A. Electropherograms of homology arms F1 and F2 (M: Marker). B. Electropherogram of total homologous fragment F (M: Marker). C. Sequence alignment. D. PCR identification of clones(-:Negative control; +:Positive control; B:Blank control; 1-8:PCR identification results of mutant strains; M: Marker).
2.3 ncRNA617回补菌株和过表达菌株的构建
以伤寒沙门菌野生菌株基因组DNA为模板,用引物PF/PR进行PCR,获得ncRNA617回补片段,该片段长270 bp(见图3A)。将ncRNA617回补片段克隆至质粒pBAD33,提取重组质粒,进行PCR和酶切双重验证,以重组质粒为模板,引物PA/PB进行PCR扩增,得到270 bp的目的带,重组质粒经双酶切也产生相同的目的带(见图3B)。将质粒送至苏州鸿讯生物科技有限公司进行测序,比对测序结果与正确序列,发现质粒中插入的序列完全正确(见图3C),表明重组质粒构建成功。提取重组质粒电转至ncRNA617缺陷菌株和野生菌株中,分别挑取6个单克隆用引物pBAD33YZ F/R进行PCR验证。12个单克隆均扩增出525 bp的条带(见图3D),表明ncRNA617回补菌株和过表达菌株构建成功。
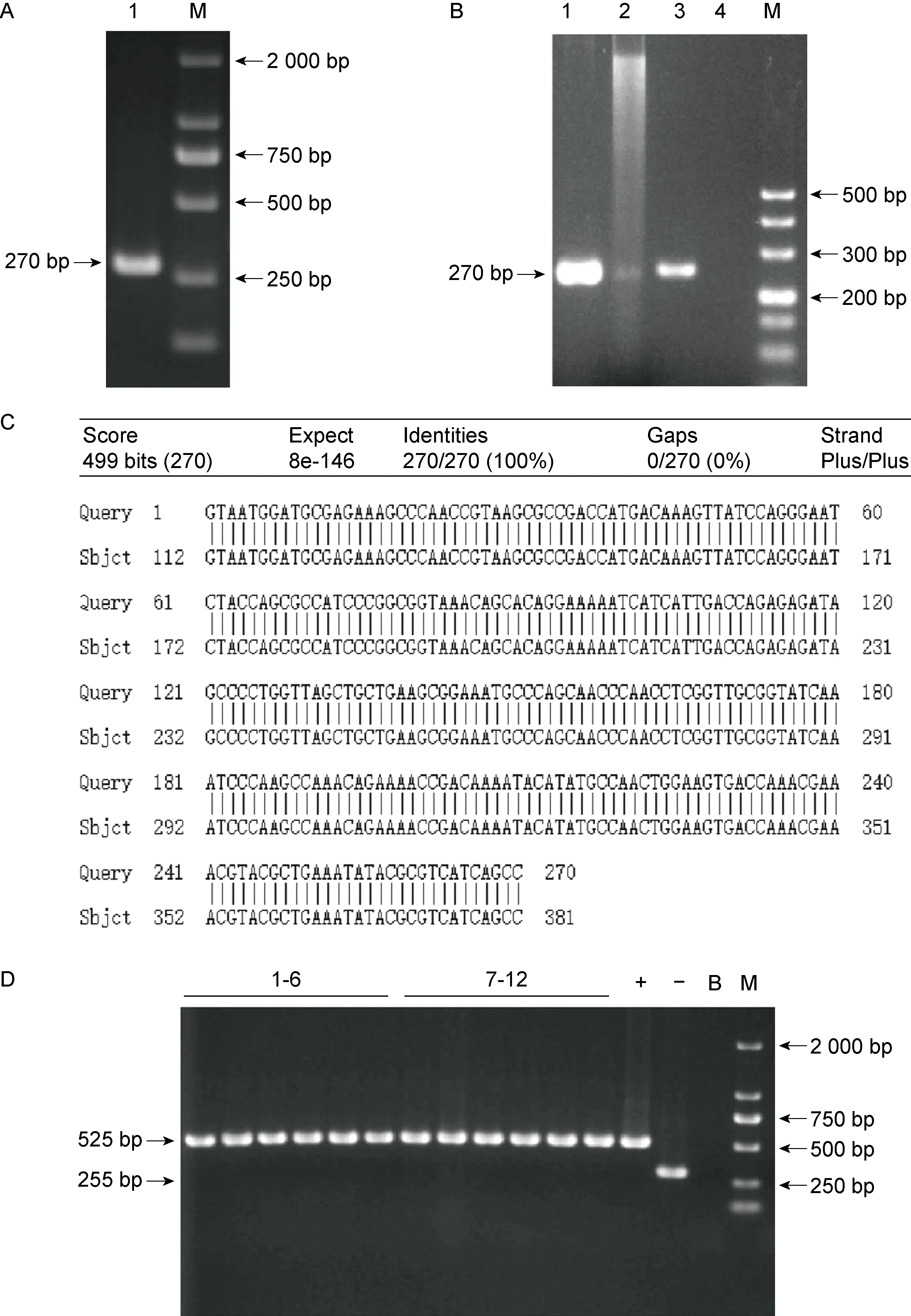
A. Amplification of complemented fragment (M: Marker). B. Validation by PCR and enzyme-cutting (1: PCR product using recombinant plasmid as template; 2: Enzyme digestion product of recombinant plasmid; 3: Positive control; 4: Blank control; M: DL500 DNA Marker). C. Blast analysis of the sequencing result of the recombinant vector. D.PCR verification of complemented strain and overexpression strain(-:Negative control;+:Positive control;B:Blank control;1-6:PCR identification results of complemented strains; 7-12:PCR identification results of overexpression strains;M: Marker).
2.4 野生对照菌株和ncRNA617缺陷对照菌株的构建
提取空质粒pBAD33分别电转至野生菌株和缺陷菌株中,分别挑取3个单克隆用引物pBAD33YZ F/R进行PCR验证,6个单克隆菌落均扩增出255 bp的条带(见图4),代表野生对照菌株和ncRNA617缺陷对照菌株构建成功。

+: Positive control; B: Blank control; 1-6: PCR products of monoclonal colonies; M: DL2000 DNA Marker.
2.5 ncRNA617可削弱伤寒沙门菌的生物膜形成能力
培养野生对照菌株(007-pBAD33)、ncRNA617缺陷对照菌株(Δ617-pBAD33)和ncRNA617回补菌株(Δ617-c617),进行生物膜形成检测(见图5A、5C),菌株Δ617-pBAD33的生物膜形成较于菌株007-pBAD33和Δ617-c617明显增强,且菌株Δ617-c617的生物膜形成能力恢复至菌株007-pBAD33的水平,表明ncRNA617缺失可使S. Typhi野生株的生物膜形成能力增强。为进一步验证ncRNA617发挥的作用,培养野生对照菌株(007-pBAD33)和ncRNA617过表达菌株(007-p617)进行生物膜形成实验(见图5B、5D),菌株007-p617生物膜形成能力较菌株007-pBAD33有明显减弱,表明ncRNA617可削弱伤寒沙门菌生物膜的形成能力。
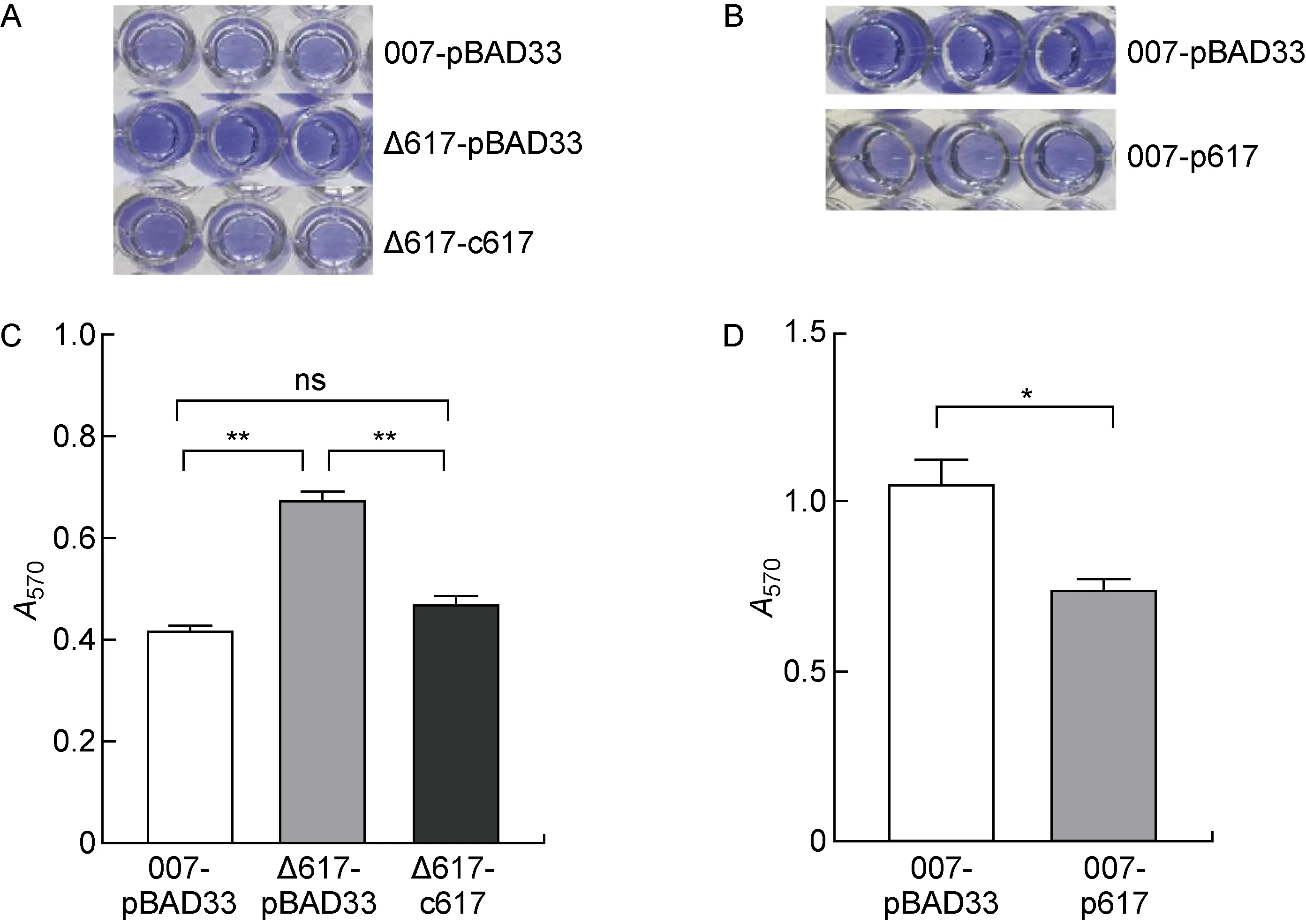
A, B:Crystal violet staining of biofilms. C, D:Columnar statistical analysis of the biofilm biomasses (**P<0.01, *P<0.05).
2.6 ncRNA617下调生物膜相关基因的表达水平
qPCR分析生物膜相关基因的mRNA水平(见图6),与野生对照菌株相比,ncRNA617缺陷对照菌株中csgD、fimA、flhD、fljB、bapA、yihP的mRNA的表达均有不同程度的上调,且差异具有统计学意义。表明ncRNA617可能是通过下调这些生物膜形成相关基因的转录表达,进而负调控伤寒沙门菌的生物膜形成能力。

**P<0.01, *P<0.05。
2.7 ncRNA617与相关基因mRNA的相互作用区域
经生物信息学软件分析发现,ncRNA617与受调控的生物膜相关基因的mRNA均有不同位置片段的结合区域(见图7),其中ncRNA617与csgDmRNA、fimAmRNA和yihPmRNA有多个不连续的碱基互补配对区域,而与flhDmRNA、fljBmRNA 和bapAmRNA 存在连续的碱基互补配对。

图7 ncRNA617与相关基因mRNA的相互作用Fig.7 Interaction between ncRNA617 and related gene mRNA
3 讨论
生物膜多细胞结构的形成属于动态进程,其中非编码RNA发挥着重要作用[16]。非编码RNA是一种只在生物体内转录而不翻译的RNA,几乎所有细菌体内都含有ncRNA,其在某些特定环境中可被激活并参与调控细菌基因的表达[17-18]。它们通常与靶mRNA进行碱基配对或者直接结合蛋白,从而在转录水平、转录后水平以及翻译水平发挥作用[19-20]。本研究前期通过RNA-Seq技术获得了生物膜状态和浮游状态的S. Typhi转录组数据,并对其中的ncRNA617进行了分子鉴定,探究其在生物膜形成过程中发挥的作用,为后续机制研究提供基础。
目前,RNA-Seq是发掘ncRNA的主要技术,许多分子生物学实验均可验证及检测ncRNA的表达,比如RT-PCR、Northern blot、微阵列杂交等方法,但是通常情况下Northern blot的特异性较高,而且该方法可检测出目的片段的大小以及是否有剪切体存在,所以从特定角度来看Northern blot是检测RNA水平的金标准[21]。本研究中,Northern blot结果提示伤寒沙门菌中确有ncRNA617的表达,长度约300 nt。为了探究ncRNA617的全长序列,本研究采用5’RACE和3’RT-PCR实验分析ncRNA617的转录起始位点和终止位点,发现ncRNA617起始位点位于mig-14终止密码子下游967 nt处,终止位点位于t2681起始密码子上游 2 378~2 560 nt处。综合Northern blot、5’RACE和3’RT-PCR的实验结果,可知ncRNA617的分子全长约270~452 nt。
为探究ncRNA617在生物膜形成过程中发挥的作用,本研究构建了野生对照菌株、ncRNA617缺陷对照菌株、回补菌株和过表达菌株,通过生物膜形成实验,发现ncRNA617可削弱伤寒沙门菌生物膜的形成能力。随后,通过qPCR实验发现,ncRNA617的缺失可使得菌毛相关基因csgD、fimA,鞭毛相关基因flhD、fljB,表面蛋白bapA,胞外多糖相关基因yihP的mRNA水平均有不同程度的上调,这提示ncRNA617可能是通过调节相关基因的表达和基质合成进而负调控生物膜的形成。
csgD是负责编码生物膜中央调控因子CsgD的基因,其在细菌稳态期细胞中表达水平会升高,不仅能促进curli菌毛和纤维素的生成[22],而且其转录过程会受诸多非编码RNA的调控,比如McaS、RprA等已知非编码RNA[23]。本研究的qPCR结果提示,ncRNA617可负调控csgDmRNA的水平,同时推测fimAmRNA水平的上调可能是由ncRNA617缺陷后引起的csgDmRNA水平上升导致,但也不排除fimA是由ncRNA617直接调控。细菌的鞭毛组成较为复杂且鞭毛基因的表达呈级联调控方式,但这种级联调控并非绝对,因为这些基因的表达除受自身调控因子的影响外,还受外界环境变化的调控[24]。flhD位于flhDC操纵子,其编码产物是鞭毛基因的主要调节子FlhDC,FlhDC负责调节二级基因启动子的活性。fliA属于二级鞭毛基因,可调控三级鞭毛基因的表达。fljB编码的鞭毛蛋白是细菌鞭毛的关键组成部分,用于鞭毛丝的形成[25]。本研究的qPCR结果提示,ncRNA617缺失后flhD和fljB的mRNA的水平升高,说明ncRNA617可能通过抑制细菌鞭毛的形成进而负调控生物膜的形成。bapA的序列在沙门菌中高度保守,其负责编码BapA蛋白,该蛋白不仅与纤维素和菌毛一起构成生物膜,还改善宿主外细菌的增殖,并增强了细菌对消毒剂和干燥剂的抵抗力[15]。本研究的qPCR结果提示,ncRNA617缺失后bapAmRNA水平升高,说明ncRNA617可影响bapA的转录水平,但是否影响其翻译水平有待进一步研究。
本研究对可能受ncRNA617调控的生物膜基质的相关基因进行了初步探索,但是ncRNA617对相关基因mRNA的影响机制目前还不明确。由于ncRNA与靶标主要通过形成互补配对来发挥调控作用,为了明确这种相互作用机制,本研究采用IntaRNA这一生物信息学工具对非编码RNA和靶基因的mRNA结合进行了预测。结果发现,ncRNA617与csgD、fimA、flhD、fljB、bapA、yihP的mRNA均有不同位置和不同长度的结合。其中,ncRNA617与csgD、fimA和yihP的mRNA有不连续的碱基配对,这点符合反式编码RNA的作用特点。但ncRNA617下调这些基因的mRNA水平是否由互补配对而引起有待深入研究。
综上所述,本研究对新发现的非编码RNA ncRNA617进行了分子鉴定,其起始位点位于mig-14 终止密码子下游967 nt处,终止位点位于t2681起始密码子上游 2 378~2 560 nt处,全长约270~452 nt。ncRNA617可能通过靶向结合生物膜形成相关基因序列从而下调基因表达,负向调控伤寒沙门菌生物膜的生成,但该调控作用的具体分子机制有待进一步研究。
