硅基电子气去除甲基氯硅烷的分子动力学模拟
2022-08-29李艳平严大洲杨涛温国胜韩治成
李艳平,严大洲,杨涛,温国胜,韩治成
(1 中国恩菲工程技术有限公司,北京 100038;2 硅基材料制备技术国家工程研究中心,河南洛阳 471023)
随着信息产业的飞速发展,人们对光纤领域产生了浓厚的兴趣。光纤通信技术因具有通信容量大、传输损耗低、保密性能好等优点,成为一种极有前途的多路通信手段。目前主要利用硅基电子气SiCl经化学气相沉积法制备光纤预制棒,但是SiCl纯度直接影响光纤原料品质,其中含金属杂质和含氢杂质因对光子产生很大的振动吸收而增加了光纤传输中光的吸收损耗。在金属硅与HCl气体反应制备SiCl的工艺过程中,金属硅中含有的少量碳为反应提供了甲基源,因而同时产生了副产物甲基氯硅烷,如CHClSiCl、CHClSiCl和CHClSiCl。光纤用高纯SiCl要求其中甲基氯硅烷含量降低到5mg/kg 或者更低。虽然国内个别生产厂家在SiCl分离提纯的核心技术方面已有一定突破,但与国外先进水平相比仍有一定的差距。
光纤用高纯SiCl对纯度的要求极高,特别是含氢杂质。因SiCl和甲基氯硅烷的沸点较接近(沸点差为9℃),很难通过传统的精馏方法完全去除甲基氯硅烷杂质。目前,SiCl的分离提纯工艺主要包括精馏法、吸附法、吸附-精馏法、光氯化法和等离子法等。其中,精馏法对含氢杂质的去除较为有效;吸附-精馏法虽然在去除金属杂质和含氢杂质时均较为有效,但存在引入新杂质和操作稳定性差的问题;光氯化法通过结合光氯化反应和精馏的方式提纯SiCl,能够避免其他提纯方法存在的问题,是目前制备光纤用高纯SiCl最合适的方法。
光氯化法实验过程中,氯气分子在紫外光照射下发生自由基链式反应生成氯自由基,氯自由基与甲基氯硅烷等含氢杂质发生化学反应,形成与SiCl具有较大沸点差的产物。光氯化反应涉及的反应为式(1)~式(4)。
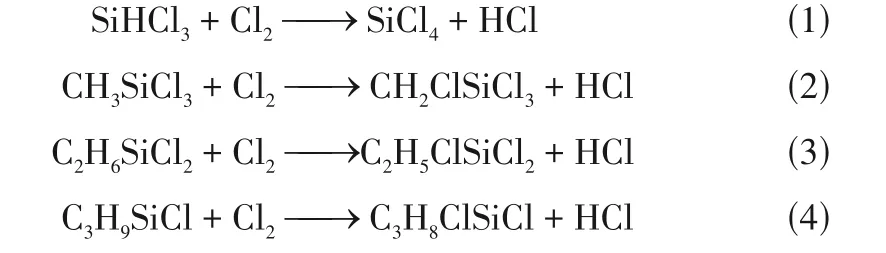
光氯化反应为自由基链式反应,副反应的发生导致相对较难控制氯化位点和氯化深度,同时高度放热的反应属性增加了反应热控制难度,当高反应热存在时很容易发生过度氯化以及其他副反应。光照强度和光照效果将会直接影响反应效果,当反应器内部光照分布不均匀时,容易在反应器黑暗处发生副反应。为了提高光氯化反应的除杂效果,进一步强化气液传热和传质效果,实现对光照和温度的有效控制,迫切需要在分子尺度上深入研究光氯化反应过程机理。
分子模拟利用计算机以原子水平的分子模型模拟分子结构和分子行为,从而获得分子体系的各种物理和化学特性。分子模拟既可从分子角度考察物理现象和物理过程(如经典分子动力学、分子力学),也可从电子水平研究化学反应路径、过渡态、反应机理等实验方法无法深入研究的过程(如量子力学、量子化学),已成为研究生物分子体系、聚合物、金属与非金属材料相关过程微观机理的有力工具。量子化学计算(quantum mechanics,QM)方法主要针对具体的分子体系,通过精确或者近似求解薛定谔方程获得体系电子水平的描述,可提供分子性质和相互作用的定量信息,并且可深入了解不能完全从实验手段获得的化学反应机理。但是QM方法计算价格高昂,所能计算的模拟体系规模偏小(约100 个原子),需要人为地假定或推测反应机理然后再加以验证,不适合用来研究涉及较多复杂反应的光氯化反应过程机理。基于经典力场的分子动力学(molecular dynamics, MD)方法在模拟过程中保持原子间成键关系及原子部分电荷不变,不能考察所模拟的分子体系中化学键的断裂和生成,无法模拟包含化学反应的分子体系行为,同样不适合用于模拟研究光氯化反应过程机理。
反应分子动力学ReaxFF MD (reactive force field molecular dynamics)在QM 和MD 方法之间架起一座桥梁,是一种将化学反应力场ReaxFF 与分子动力学MD相结合的计算方法。基于键级的化学反应力场ReaxFF,全面考虑了键、键角、二面角、共轭、氢键以及范德华力和库仑相互作用,且通过电负性平衡算法(EEM)动态更新体系的部分电荷,能够在不预先定义化学反应路径的前提下模拟分子体系的化学反应。ReaxFF MD已被广泛地应用于各种材料体系,包括高能物质爆炸反应、裂解和燃烧反应、晶体及晶体表面相互作用等。
本文采用ReaxFF MD 方法模拟研究硅基电子气SiCl中一种主要的氯硅烷杂质甲基氯硅烷(CHSiCl)的光氯化反应过程机理,基于化学反应信息分析和可视化方法,获得主要反应中间体和基元反应,确定反应物CHSiCl的主要转化路径,定量地比较反应物Cl分子和氯自由基(Cl·)对CHSiCl消除效果的影响,并研究了光氯化反应适宜的反应温度。由此获得的模拟研究结果能够为进一步优化硅基电子气除杂的光氯化反应过程工艺提供理论支撑。
1 反应模型构建及模拟策略
反应模型构建时,首先对单个CHSiCl、Cl分子及Cl·进行分子结构优化,然后将100 个CHSiCl分子与100 个Cl分子(CHSiCl&Cl反应模型) 或者100 个CHSiCl分子与200 个Cl·(CHSiCl&Cl·反应模型)按照0.0001g/cm密度随机地放置在一个长度为715.2Å(1Å=0.1nm)的立方型模拟盒子中。最后,采用universal力场对反应模型中分子分布基于能量最小化进行结构优化,最终构建的CHSiCl&Cl反应模型如图1 所示。模拟过程中,周期性边界条件()应用于立方型模拟盒子的三个方向上,且采用正则系综(NVT)、Berendsen 控温方法(温度的阻尼常数为0.02ps)及0.2fs 的模拟时间步长。基于模拟策略设计相关算例中主要参数如表1所示。

图1 模拟体系CH3SiCl3&Cl2的反应模型
表1 模拟算例中主要参数
模拟计算开始后,反应体系首先在300K 弛豫50ps,然后从300K 升温到目标反应温度并在目标温度恒温反应1000ps。为了探索适宜的光氯化反应温度,并考虑到Cl的解离能(要求反应温度需要大于100℃),反应温度分别设置为323K、373K及423K。模拟过程中形成的中间产物将停留在反应体系,以便进行更深层次的化学反应。每间隔1000 时间步(0.2ps)记录一次每个分子的运动轨迹及键级相关数据等。所有算例均采用LAMMPS代码进行计算,并借助VARxMD(visualization and analysis of reactive molecular dynamics)软件进行复杂的化学反应信息分析。
2 模拟结果
2.1 模拟参数合理性验证
首先,模拟计算过程中采用的分子反应动力学力场已成功应用于模拟研究采用甲基氯硅烷前体生长SiC 的绝热反应动力学,该反应力场已经对本文所涉及的不同类型化学键和化学反应参数进行了优化,验证了模拟所应用反应力场参数准确性和可靠性。其次,参照已发表文献并结合模拟需求综合考虑后设定模拟计算过程中应用的主要模拟参数,包括动力学模拟时间、模拟时间步长及模拟盒子尺寸等。在一些同样应用ReaxFF MD 方法的模拟研究中,模拟时间步长分别设置为0.1fs和0.2fs,动力学反应时间分别为1000ps、5ps、12.5ps和750ps。在高温下为了有效覆盖相空间和平稳地发生碰撞和反应,一般采用较小的时间步长0.1fs,而本文的反应温度(323K、373K、423K)较低,将模拟时间步长设置为0.2fs 可满足计算精度。一般来说,模拟时间步长应当比耗时最短的分子或原子运动小一个数量级或者接近0.5~1.0fs。由此可见,本文中应用的动力学模拟时间、模拟时间步长等模拟参数及反应力场参数均合理。
为测试模拟盒子尺寸依赖性,同时在2倍尺寸模拟盒子和4 倍尺寸模拟盒子中进行了模拟计算,模拟体系包含原子数由1000 个分别增加到2000 个和4000 个。以模拟体系CHSiCl&Cl为例(图2),模拟盒子尺寸增加并不会改变反应物CHSiCl分子数量随着模拟体系演化而逐渐减少的变化趋势。同样,其数量变化速率随着模拟体系演化均在零值附近波动,也没有受到模拟盒子尺寸的影响。在模拟体系CHSiCl&Cl·中,同样可观察到类似的动态变化趋势。此外,如图3所示,模拟体系CHSiCl&Cl与CHSiCl&Cl·在不同尺寸的模拟盒子中演化相同时间反应物CHSiCl分子的转化率基本一致。这些结果一方面表示反应物CHSiCl分子在模拟体系演化过程中主要参与可逆化学反应,反应物CHSiCl分子在体系演化过程中交替进行生成反应和转化反应,但因转化量远大于生成量,使得反应物分子数量总体上呈现出逐渐减小的变化趋势,这一变化过程也符合实际反应过程,说明了模拟结果的合理性。另一方面说明模拟盒子尺寸并不会影响反应物CHSiCl分子随着模拟体系演化的动态变化趋势及模拟体系演化相同时间获得的转化率,验证了本文采用模拟盒子尺寸的合理性。
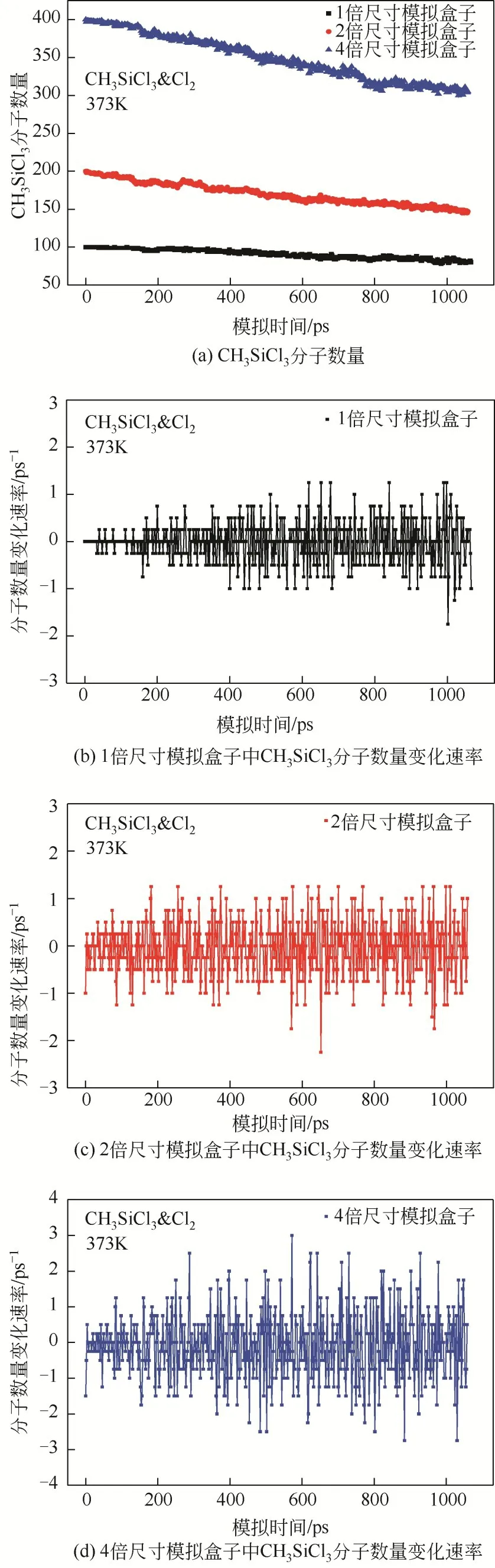
图2 模拟体系CH3SiCl3&Cl2在不同尺寸的模拟盒子演化过程中(373K),反应物CH3SiCl3分子数量和变化速率的动态变化趋势
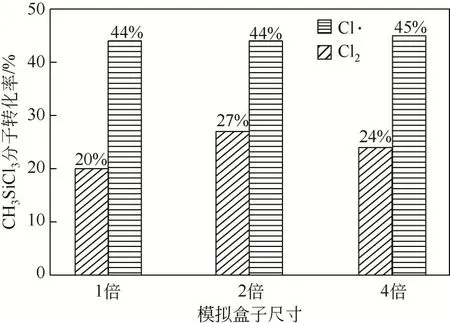
图3 模拟体系CH3SiCl3&Cl2与CH3SiCl3&Cl·在不同尺寸的模拟盒子中演化相同时间后(373K)反应物CH3SiCl3分子转化率
为验证动力学模拟时间合理性,设计了将动力学模拟时间从1ns 分别延长到2ns 和3ns 的模拟算例。对比分析模拟体系演化过程中形成的主要中间产物分布(表2、表3 和表6)可知,模拟体系CHSiCl&Cl的主要中间产物分布在不同演化时间下产生了大部分重叠,只是在出现频次上存在差异,说明主要中间产物分布基本上没有受到模拟时间的影响。同样,对比分析主要中间产物分布可知,在模拟体系CHSiCl&Cl·中能够观察到相同的现象。因此,延长动力学模拟时间并不会明显改变模拟体系内主要中间产物分布,反应体系在演化时间小于1000ps 时已达到反应平衡,说明动力学模拟时间1000ps 的合理性。此外,本研究以获得模拟体系达到平衡后发生的基元反应为研究目的,延长动力学模拟时间虽然可以提高反应物分子转化率,但同时也将大大增加物理计算时间,需要权衡计算成本和反应效率匹配问题。例如,当均采用4 个计算核心对反应体系CHSiCl&Cl进行模拟计算时,反应体系演化1000ps、2000ps、3000ps、10000ps 所需要的物理计算时间分别为361min、2952min、5522min 和36228min。由此可见,物理计算成本将因动力学模拟时间延长而急剧增加,而计算获得的有效化学反应信息并不因动力学模拟时间延长而增多。因此,将动力学模拟时间设置为1000ps 既可达到研究目的又可获得合理模拟结果,同时还可节省物理计算时间。

表2 模拟体系CH3SiCl3&Cl2反应演化2000ps过程中形成的主要中间产物(373K)
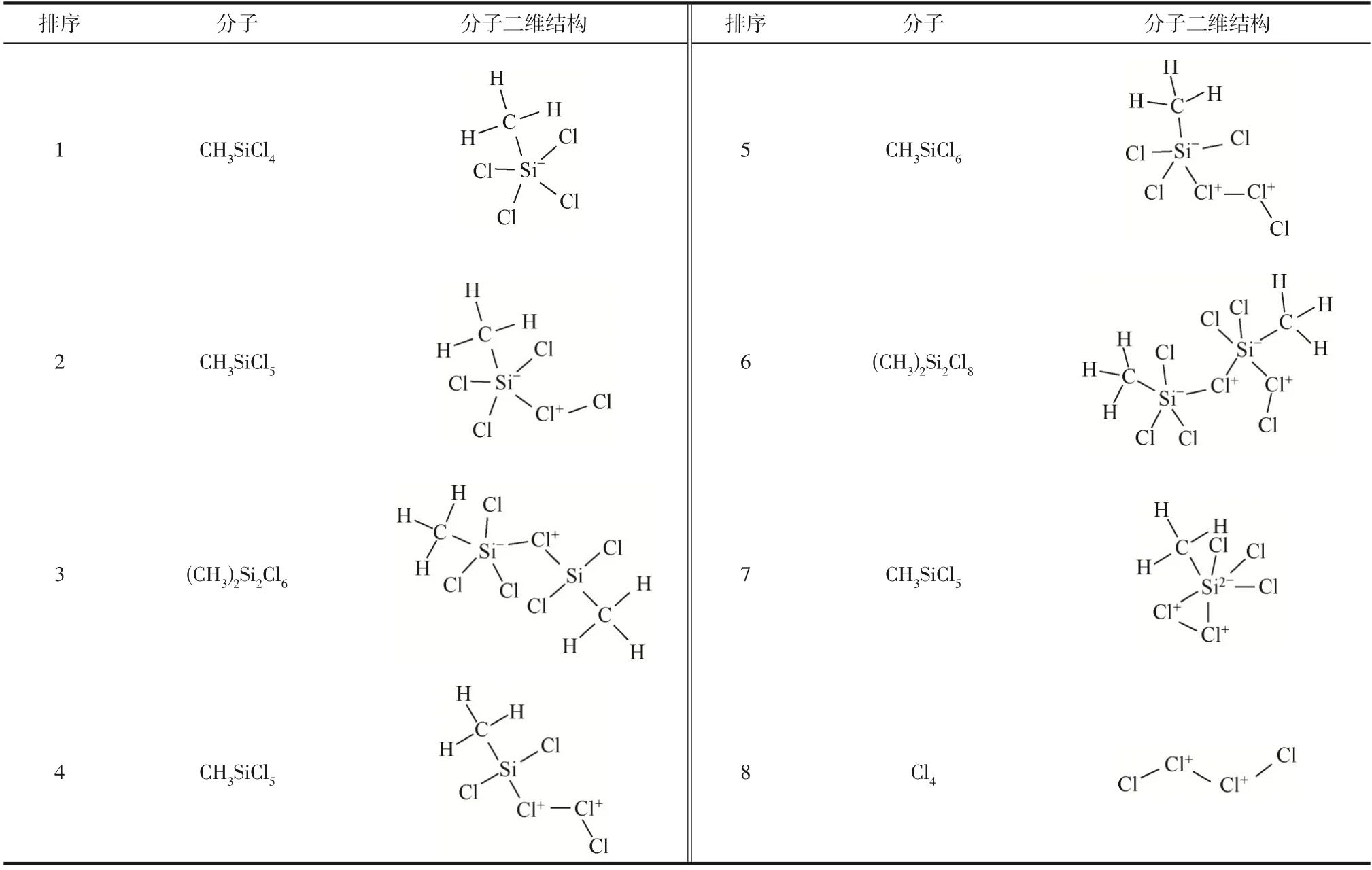
表3 模拟体系CH3SiCl3&Cl2反应演化3000ps过程中形成的主要中间产物(373K)
为验证模拟时间步长的合理性,将模拟时间步长减小为0.1fs进行计算。模拟体系CHSiCl&Cl和CHSiCl&Cl·在373K 与1 倍尺寸模拟盒子中演化1000ps 后获得主要中间产物分布分别如表4 和表5所示。分别对比表4与表5、表6与表7可知,模拟时间步长减小为0.1fs 后得到的主要中间产物分布与应用0.2fs 模拟时间步长相比基本一致,只是在分子出现频次上存在微小差异。这些结果说明文中应用的0.2fs 模拟时间步长具备合理性,可满足计算精度。
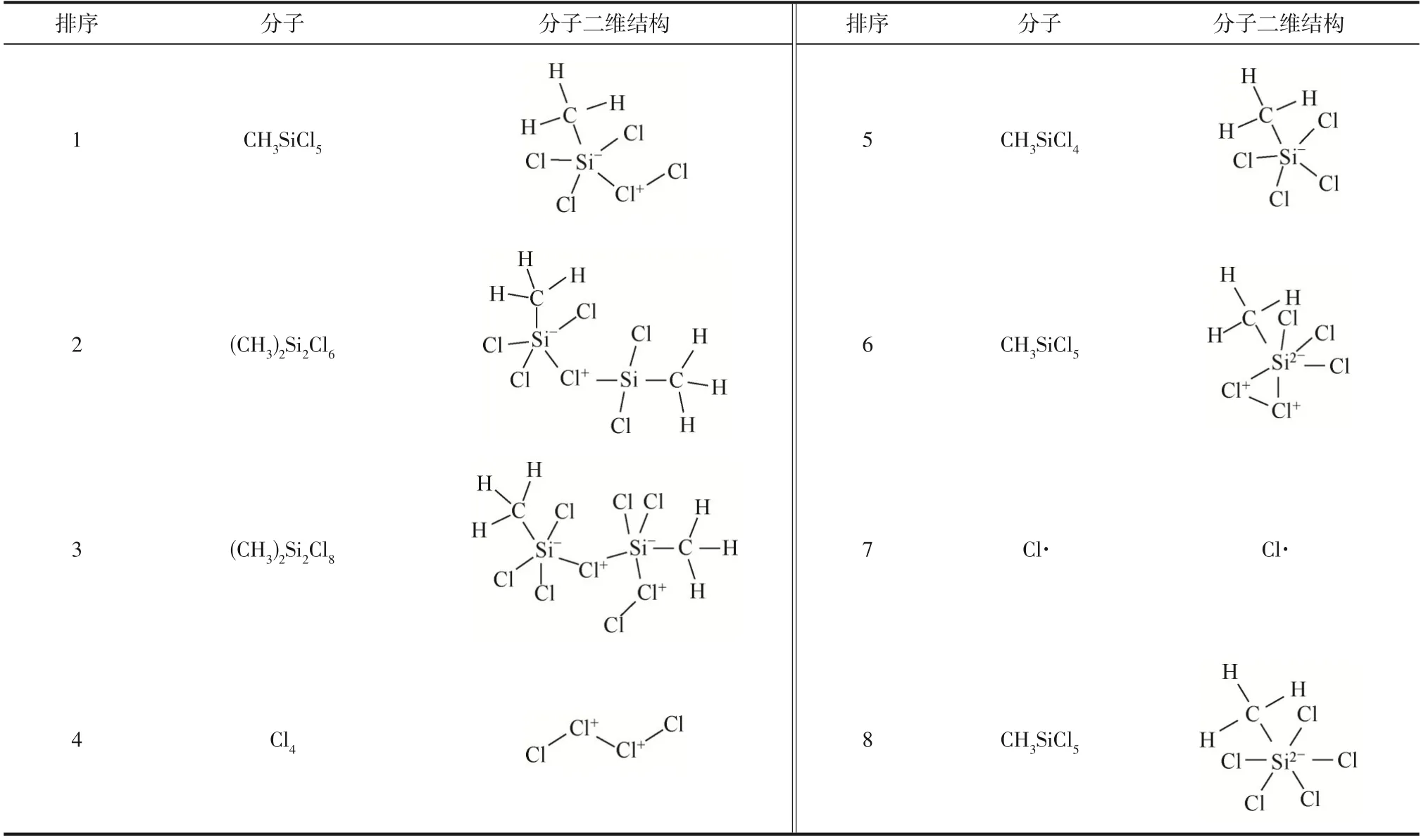
表4 模拟体系CH3SiCl3&Cl2反应演化1000ps过程中形成的主要中间产物(0.1fs,373K)
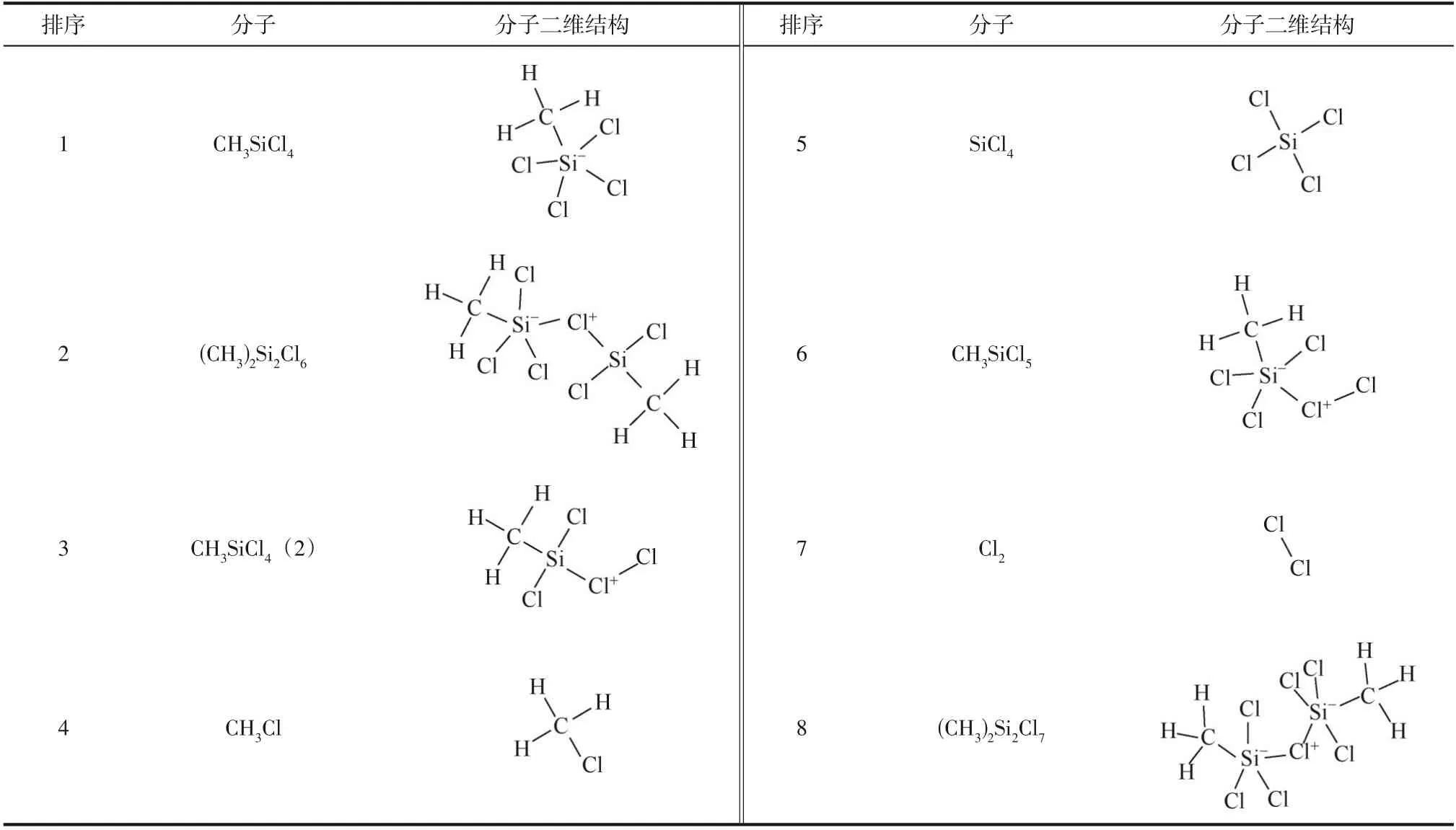
表5 模拟体系CH3SiCl3&Cl·反应演化1000ps过程中形成的主要中间产物(0.1fs,373K)
综上所述,无论是分析已获得模拟结果还是对比已发表研究中相关参数,本文中所采用的反应力场参数、模拟盒子尺寸、动力学模拟时间及模拟步长均合理。
2.2 模拟体系CH3SiCl3&Cl2中的主要中间产物
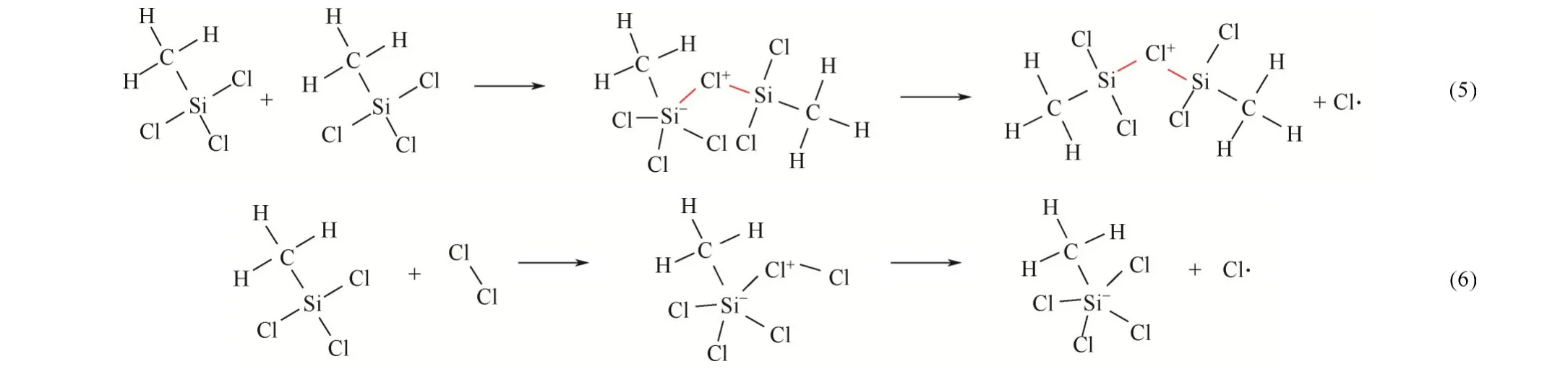
模拟体系CHSiCl&Cl在373K经过1000ps(算例Ⅱ)恒温反应演化共产生了17 种中间产物,除反应物CHSiCl和Cl分子外,演化过程形成的主要中间产物分子式及结构式如表6所示。显然,体系演化过程中产生数量最多的中间产物是CHSiCl分子中Cl原子与Cl分子成键形成的CHSiCl分子。然而,CHSiCl分子的稳定性差,分子中Cl—Cl 键容易断裂形成CHSiCl分子。此外,模拟体系中较多的反应物Cl分子间易通过形成Cl—Cl 键而产生链状Cl分子。与此类似,模拟体系中较多的反应物CHSiCl分子间易通过Cl 原子形成Si—Cl—Si桥键进而形成(CH)SiCl分子。因为(CH)SiCl分子中1 个Si 原子与5 个原子成键降低了分子稳定性,使得Si 原子易脱去1 个Cl 原子形成稳定性增强的(CH)SiCl分子。此外,模拟体系演化过程中也可观察到较多Cl·。2 条重要的反应物分子转化路径见式(5)、式(6)。
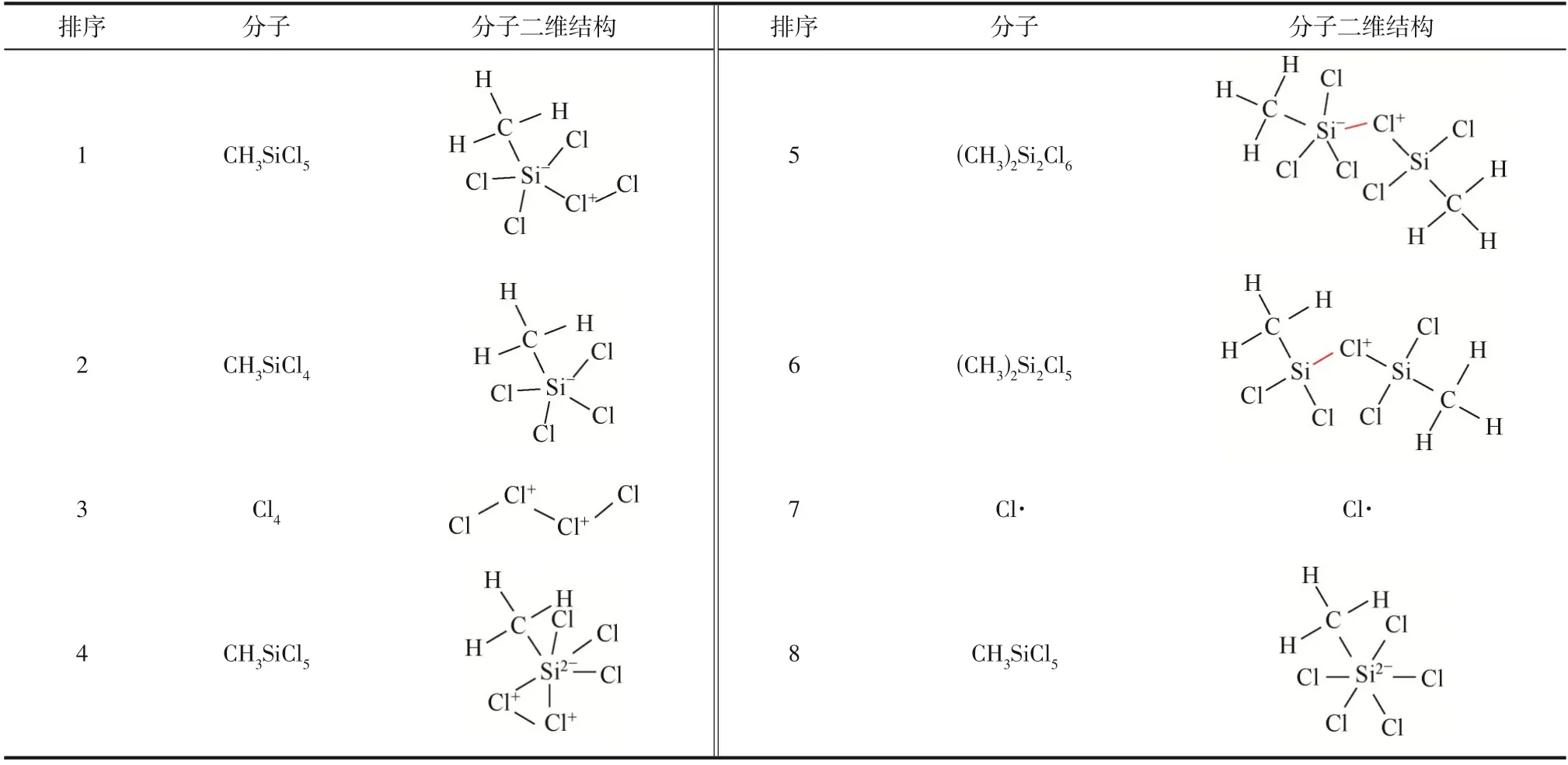
表6 模拟体系CH3SiCl3&Cl2反应演化1000ps过程中形成的主要中间产物(373K)
当模拟体系CHSiCl&Cl在473K 恒温反应时,可获得反应物CHSiCl分子中C原子脱去1个H原子形成中间产物CHSiCl。据此推测,CHSiCl分子中C—H 键的键能相对较大,只有当模拟体系的反应温度升高到一定值后,才发生C—H键的断裂。
2.2 模拟体系CH3SiCl3&Cl·中的主要中间产物
光氯化除杂反应过程中,1个Cl分子吸收光能量后将解离为2 个活泼Cl·参与反应过程,因此有必要对模拟体系CHSiCl&Cl·化学反应机理进行模拟研究。该体系在373K恒温反应1000ps(算例Ⅴ)过程中共产生了31 种中间产物,除去反应物CHSiCl和Cl·外,演化过程形成的主要中间产物分子式及结构式如表7所示。对比表6可知,在模拟体系中引入反应活性较高的Cl·替代反应活性相对较低的Cl后,模拟体系CHSiCl&Cl·演化过程中出现频次最高的中间产物为CHSiCl分子,该分子由反应物CHSiCl分子受到活泼Cl·攻击形成1个Si原子与Cl原子键合而形成。表7中出现频次最高的CHSiCl分子对应表6 中出现频次较高的CHSiCl分子,这两个分子都是因反应物Cl·/Cl分子与反应物CHSiCl分子直接相遇并发生反应生成的中间产物。出现频次排第2位的中间产物为2个反应物CHSiCl分子通过Si—Cl—Si 桥键形成的(CH)SiCl分子。实际上,含Si原子的分子易通过Si—Cl—Si 桥键结合形成大分子,比如出现频次排第4位的(CH)SiCl分子。与CHSiCl&Cl模拟体系相比,明显的差异是在CHSiCl&Cl·体系中因Cl·引入产生了出现频次较高的中间产物CHCl和SiCl分子,这一结果说明因反应体系内较多Cl·的存在,使得反应物CHSiCl分子中C—Si 化学键发生断裂,进而促进CHCl和SiCl分子的形成[式(7)]。
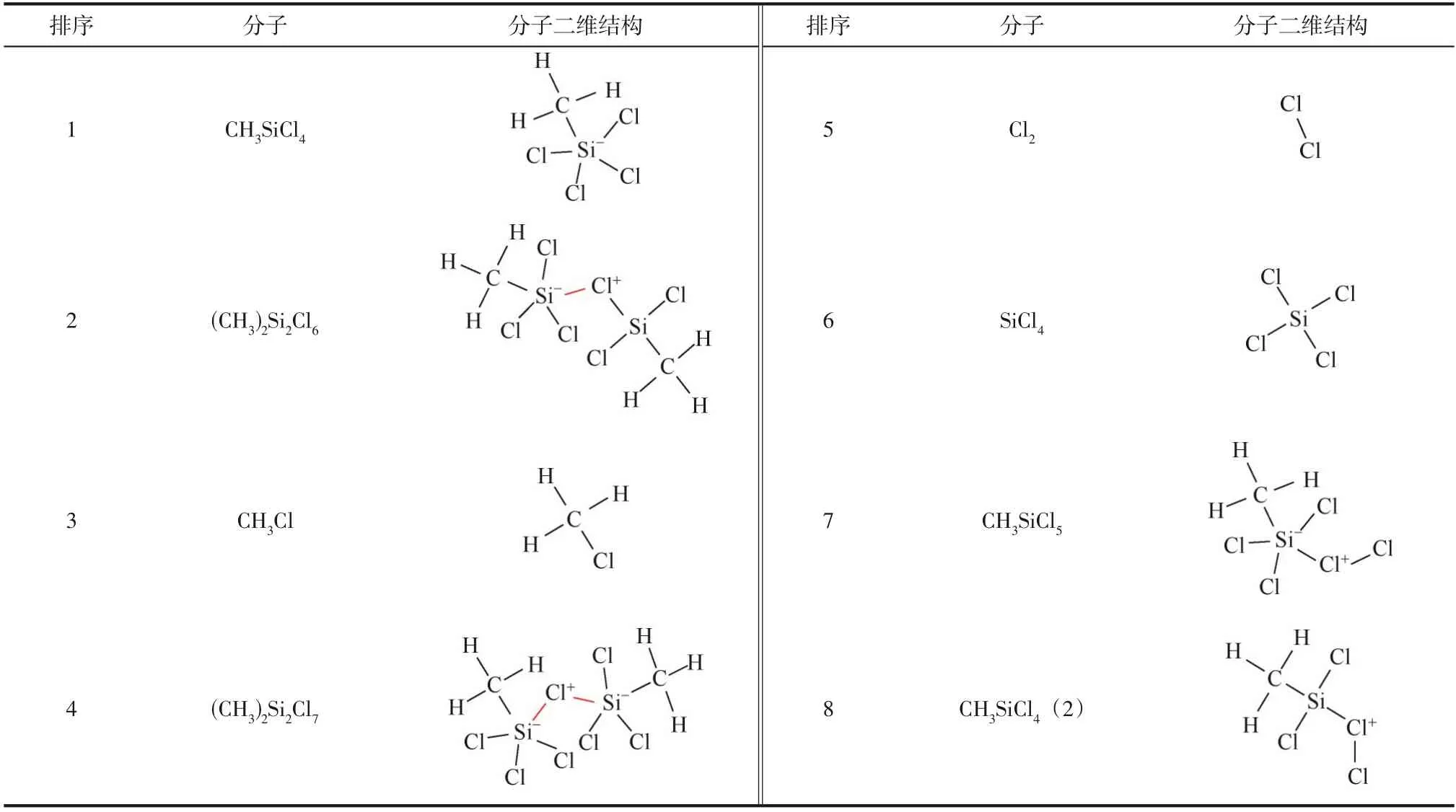
表7 模拟体系CH3SiCl3&Cl·反应演化1000ps过程中形成的主要中间产物(373K)

由此可见,通过控制硅基电子气SiCl光氯化反应过程实验条件,不仅可以消除痕量杂质CHSiCl,还可以将CHSiCl转化为SiCl。在光氯化反应脱除三氯氢硅中甲基氯硅烷实验中同样可观察到生成SiCl分子,与模拟结果一致。模拟体系内活泼Cl·也可键合为Cl分子,但是反应演化过程中不再形成类似于Cl的长链分子。此外,模拟体系演化过程中,出现频次最高的中间产物CHSiCl及反应物CHSiCl分子因受到活泼Cl·进攻而分别形成中间产物CHSiCl与CHSiCl(2),这两个分子的共同点是均形成Cl—Cl化学键。
2.3 反应物Cl2与Cl·对CH3SiCl3消除效果的对比分析
固定模拟体系CHSiCl&Cl与CHSiCl&Cl·反应温度均为373K 并且CHSiCl分子初始数量均为100 个,定量比较反应物Cl与Cl·对杂质分子CHSiCl的消除效果。如图4所示,反应物CHSiCl分子数量随着模拟体系演化逐渐减小,在相同的反应时间段范围内,模拟体系CHSiCl&Cl·中CHSiCl分子的消耗数量和消耗速率均明显大于模拟体系CHSiCl&Cl。实际上,模拟体系CHSiCl&Cl·与CHSiCl&Cl演化相同时间后,CHSiCl分子转化率可分别达到44%和20%,而反应物Cl分子对CHSiCl的去除效果只能达到同样条件下Cl·对CHSiCl去除效果的45%。由此可见,光氯化反应过程中通过光照将Cl分子解离为Cl·确实可以提高硅基电子气中痕量杂质甲基氯硅烷的去除效果。

图4 模拟体系CH3SiCl3&Cl2与CH3SiCl3&Cl·演化过程中(373K)反应物CH3SiCl3分子数量动态变化趋势
2.4 反应温度对CH3SiCl3去除效果的影响
模拟体系演化过程中,反应物CHSiCl分子转化率受到反应温度的影响。当模拟体系CHSiCl&Cl与CHSiCl&Cl·在不同的反应温度演化时,反应物CHSiCl分子数量动态变化情况分别如图5 和图6所示。当模拟体系CHSiCl&Cl分别在323K 和373K反应演化时,反应物CHSiCl分子数量随着模拟体系演化呈现逐渐下降的变化趋势,并且温度越高该下降趋势越明显。但是当反应温度继续升高到423K后,CHSiCl分子数量随着模拟体系演化的下降趋势明显变缓,甚至在反应后期出现了在某平衡值附近波动的趋势。由此可见,当反应温度达到373K 后,通过继续增加反应温度的方式并不能明显提高CHSiCl分子的去除效果,这可能是因为反应温度继续提高后增加了某些相关中间产物转化为CHSiCl分子的基元反应的发生概率。
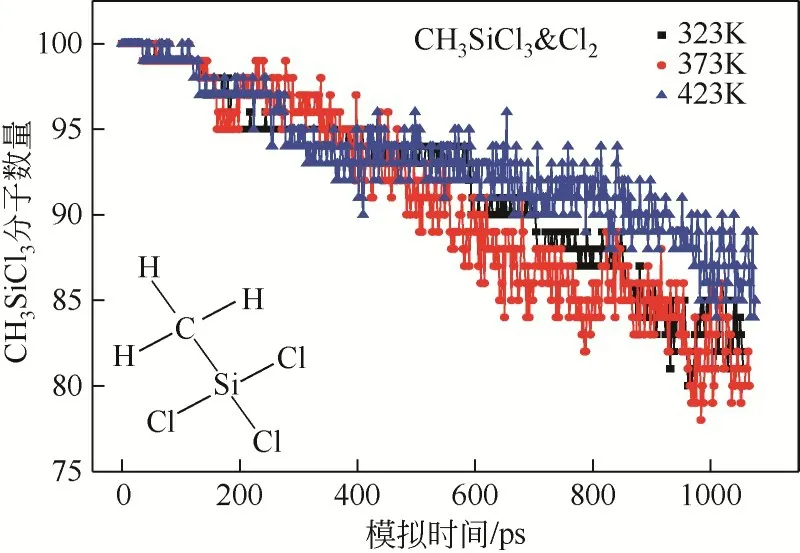
图5 模拟体系CH3SiCl3&Cl2在不同的反应温度演化过程中,反应物CH3SiCl3分子数量动态变化趋势
当模拟体系CHSiCl&Cl·在不同的反应温度演化时获得的反应物CHSiCl分子数量动态变化趋势明显不同于模拟体系CHSiCl&Cl,如图6 所示,随着模拟体系CHSiCl&Cl·演化,CHSiCl分子数量在323K、373K及423K的反应温度下均表现出了明显的下降趋势,且下降趋势的排序为423K≈373K>323K。与模拟体系CHSiCl&Cl相比,模拟体系CHSiCl&Cl·演化过程中反应物CHSiCl分子数量的消耗受到反应温度的影响比较明显,尤其是当反应温度设置为423K时。整体上看,CHSiCl分子的去除效率随着反应温度的增加而逐渐增加,但是当反应温度达到373K 后,继续提高反应温度并不能明显提高其去除效率。由此可见,去除痕量杂质甲基氯硅烷的光氯化反应存在最佳反应温度,对光氯化反应除杂工艺进行优化时需要重点关注反应温度对杂质去除效率的影响,因此需要实时检测反应体系内温度变化并及时移除反应热。
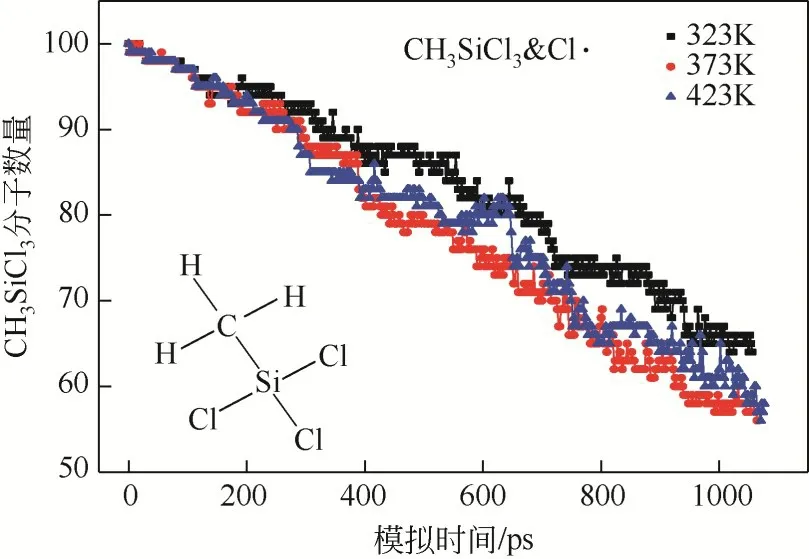
图6 模拟体系CH3SiCl3&Cl·在不同的反应温度演化过程中,反应物CH3SiCl3分子数量动态变化趋势
如图7所示,当模拟体系停止反应演化后,模拟体系CHSiCl&Cl在反应温度323K、373K 及423K时的CHSiCl分子转化率分别为19%、20%及15%,而模拟体系CHSiCl&Cl·的CHSiCl分子转化率分别为36%、44%与42%。显然,与反应物Cl分子相比,在反应体系中引入Cl·后可明显提高CHSiCl分子去除效率。

图7 模拟体系CH3SiCl3&Cl2与CH3SiCl3&Cl·在不同反应温度演化过程中反应物CH3SiCl3分子转化率
3 结论
本文针对硅基电子气去除痕量甲基氯硅烷(CHSiCl)杂质的反应过程进行了模拟研究,定量地比较了反应物Cl与Cl·及反应温度对CHSiCl分子去除效果的影响,同时探究了不同模拟体系中产生的主要中间产物及CHSiCl分子的主要转化路径。模拟结果表明,在反应体系中引入活泼自由基Cl·后可以明显地提高CHSiCl分子去除效率,在模拟的时间段内基本可提高2倍的去除效率。针对CHSiCl分子去除效率,反应温度对模拟体系CHSiCl&Cl·的影响明显大于模拟体系CHSiCl&Cl,但是反应温度与去除效率之间并非单调变化。为了使CHSiCl分子去除效率达到最大,在模拟的反应温度范围内存在一个最佳反应温度(373K)。此外,只有在CHSiCl&Cl·反应体系演化过程中,可观察到数量较多的CHSiCl分子断裂C—Si键而形成小分子中间产物CHCl及SiCl。本文所获得的模拟结果有望为进一步优化硅基电子气通过光氯化反应除去痕量甲基氯硅烷杂质的工艺过程提供基础理论支撑。
