菝葜叶绿体基因组的组装分析与分子标记研究*
2022-08-26钟浩天蒋莉萍江宇慧胡志刚刘义飞
钟浩天 ,蒋莉萍 ,江宇慧 ,胡志刚 ,余 坤 ,2,刘义飞 ,森 林 ,2**
(1. 湖北中医药大学药学院 武汉 430065;2. 湖北中医药大学教育部重点实验室 武汉 430065)
菝葜(Smilax chinaL.)又名金刚藤,为百合科菝葜属药用植物[1]。菝葜主要自然分布于海拔2000 m 以下的林下、灌丛中或山坡上,以湖北、湖南、江苏、四川等省野生资源居多[2]。目前湖北为菝葜栽培种植的主产区,相应的菝葜药材品质也具有优势[3-5]。2019年菝葜(金刚藤)被纳入了湖北省首批道地药材“一县一品”优势单品在通城县进行重点培育。通城县菝葜药材大规模种植处于国内领先地位,其种子质量标准、肥料的使用和基地生态环境等方面的技术也有重大突破[6-7]。迄今为止,全国范围内菝葜药材已产生了可观的社会、经济与生态效益,并在药材产业化发展中积累了宝贵的经验。
菝葜具有抗炎、抗肿瘤及抗菌等药理作用,可用于治疗痛风和痢疾水肿等疾病[8-10]。传统中医理论认为菝葜具有利湿去浊、祛风除痹、解毒散瘀等功效,也被运用于各种中成药当中,如金刚藤胶囊和疏风活络丸等。《中国药典》规定菝葜主要以根茎入药,为不规则块状或弯曲扁柱形,有结节状隆起,表面黄棕色或紫棕色,具圆锥状突起的茎基痕,并残留坚硬的刺状须根残基或细根[1]。另外,其同属物种光叶菝葜(俗称土茯苓)同样属于药典品种,在湖北有部分分布,作为商品时也有混淆的现象出现。
叶绿体基因组变异序列广泛用于植物的分子标记开发、分子鉴定和系统发育关系分析等研究工作。国内科研团队主要利用叶绿体基因序列,建立了适用于中草药分子鉴定的“DNA 条形码技术”[11-13]。在中药骨碎补的研究中,通过高通量测序和精细组装获得了7 种槲蕨近缘物种的叶绿体基因组全序列,进一步提出利用叶绿体基因组高变区建立超级DNA 条形码的建议[14]。高通量测序的技术进步,包括读长较短的二代技术和读长较长的三代技术[15]进一步促进了植物叶绿体基因组的组装和研究。在叶绿体基因组的组装中,短读长测序因建库缺陷所缺失的信息会降低组装精度;而长读长测序却存在相对较高的每碱基错误率(10%-15%)[16],同样限制了组装精度。而新出现的以Unicycler 软件为代表的算法[17],它针对细菌基因组[18]和植物叶绿体基因[20]的环状结构,将短读长与长读长数据结合起来组装环形基因组,可获得更加准确的结果。
目前对菝葜属植物系统发育关系相关的研究较少。Kim 等利用叶绿体基因组中rbcL等4 个基因对百合科系统发育关系进行确定,其中菝葜属与油点草属关系最近,郁金香属、猪牙花属和老鸦瓣属聚为一支,百合属与贝母属聚为一支且离菝葜属最远[19]。此外,百合科祖先物种分化时间主要位于在99.9Mya 到125.1Mya 之间[20-21],而对菝葜属的报道出现了不同的记录,其中包括古新世晚期-始新世早期(48.6-55.8 Mya)以及始新世中期(37.2-48.6 Mya)[22-23]。
本研究的材料来自湖北通城的菝葜幼嫩叶片,拟借助Nova-seq6000 及PromethlON 高通量测序平台获得充足的序列信息并探索最佳组装方案。随后对菝葜在百合科中的系统发育地位以及与分化时间进行分析,探明菝葜的正确进化分类学地位。此外与近缘物种叶绿体基因组结构进行比较分析,对其蛋白质编码基因区的不同基因所受的选择压力进行分析,进而筛选适宜的SSR 引物,为菝葜中药材资源鉴定和品质评价分析提供理论基础和方法保证。
1 材料与方法
1.1 植物材料的采集与叶绿体基因组的测序与组装
本研究中所用的菝葜植物材料来源见表1,由湖北中医药大学汪乐原教授鉴定为百合科菝葜属菝葜(S.china)。采用改良后的CTAB(十六烷基三甲基溴化铵)法对菝葜幼嫩叶片基因组DNA 进行提取,将液氮速冻后的叶片进行打粉,对粉末进行预处理,抑制脱氧核糖核酸酶活性并去除糖类及酚类等杂质,利用CTAB 溶液溶解细胞膜使核酸沉淀,通过氯仿:异戊醇(24:1)去除蛋白质杂质,吸取其上清液使其中的核酸于异丙醇中沉淀,最后利用70%乙醇和无水乙醇进行脱 色 ,得 到 DNA 母 液 。 使 用 Nova-seq6000 及PromethlON 高通量测序平台开展测序,采用fastp 软件[24]对短读长数据(short reads)进行质量控制,利用NanoFilt软件[25]对长读长数据(long reads)开展预处理。将NCBI 公开数据库中的菝葜叶绿体基因组(NC_049022.1)作为组装引导序列[26],采用 Canu 软件[27]组装长读长数据结果,并用Unicycler 软件[17]开展短读长数据独立组装和长短读长数据混合组装,将结果与NCBI参考序列进行比对,并利用Sanger 测序进行验证。所得结果提交NCBI 的 GenBank 库,获得登录号No:OL372240。

表1 本实验所用的菝葜植物材料信息
1.2 叶绿体基因组的注释
基于组装后的目标序列,使用GeSeq(https://chlorobox.mpimp-golm.mpg.de/geseq.html)软件[28]对菝葜叶绿体基因组进行注释,环形图使用Circos 软件[29]进行绘制(图1)。
1.3 系统发育分析
本文所引用数据集来自NCBI 公开数据库,参考百合科[19,30-31]和菝葜属[26]的相关研究选取来自于百合科中16 个族内的23 个属共79 个物种,并以宽叶香蒲(Typha latifoliaL.,NC_013823)、黑三棱(Sparganium stoloniferum(Graebn.) Buch. -Ham. ex Juz., NC_044634)、美人蕉(Canna indicaL.,MN832865)、粉鸟蝎尾蕉(Heliconia collinsianaGriggs,NC_020362)及穴芭蕉(Musa troglodytarumL.,NC_056833)作为外类群。以84 个物种叶绿体基因组全部完整的编码基因所编码蛋白质的氨基酸序列信息作为数据集,通过OrthoFinder(v2.5.2)软件[32]利用 MAFFT(v7.486)[33]进行多序列比对,后利用ProtTest(v3.4.2)软件[34]进行检验,AIC 与BIC 检测结果均指示HIVb+I+G 为最适替换模型,随后通过RAxML(v8.2.10)软件[35-37]构建系统发育树。并以最佳合议树作为时间尺度系统发育关系构建的起始树。
随后,基于84个物种中9个共有基因(psaA、psaB、psbA、psbB、psbC、psbD、rbcL、petA、petB)作为数据集,将RAxML 推测的最佳合议树作为起始树,利用BEAST(v2.6.4)[38]对百合科各物种的分歧时间进行考察。在分子钟假设下,为将遗传距离转化为分歧时间,至少需要一个可以提供时间信息的校正点,本实验中使用美人蕉属(Canna)、芭蕉属(Musa)和蝎尾蕉属(Heliconia)(tMRCA为 83Mya)[39]以 及 黑 三 棱 属(Sparganium)和香蒲属(Typha)(tMRCA为70Mya)[40-41]的最近共同祖先时间作为时间尺度下的系统发育树的校正点。在BEAUti中设置贝叶斯迭代模型,分子钟类型设置为Relaxed Clock Log Normal(放松对数分子钟),在BEAST 中对共84 个物种叶绿体基因组中的蛋白质编码区数据集进行迭代计算4 × 107代,其中每1000 代保留一代样本,为对数据的各项参数收敛程度进行考察,把经计算所得的结果导入Tracer(v1.7.1)软件中进行检测,确认所有参数的ESS 值均大于200,认为之前的运算已经到达收敛终点。将BEAST 计算所得的40000 棵树导入TreeAnnotator 中删去最初的10%样本,并以后验概率大于95%的部分得到合议树。
1.4 叶绿体基因组的比较分析
为了探索百合科中及菝葜属内叶绿体基因组之中的差异,本实验对菝葜属五个物种菝葜(S.china)、白背牛尾菜(S. nipponicaMiq.)、土茯苓(S.glabraRoxb.)、小叶菝葜(S. microphyllaC. H. Wright)、S.glyciphyllaJ.White、郁金香属的阿尔泰郁金香(Tulipa altaicaPall. ex Spreng)和萱草属的萱草(Hemerocallis fulva(L.)L.)等物种进行考察,其中S. glyciphylla虽然并无中文命名,但与菝葜和土茯苓亲缘关系较近,为菝葜属重要物种,故列入研究对象。通过IRscope[42]研究IR区域边界的收缩与扩张现象,并对上述物种叶绿体基因组的保守性进行分析,使用软件为mVISTA(https://genome.lbl.gov/vista/mvista/submit.shtml),设置的模式为Shuffle-LAGAN,使用的参考为NCBI 上的参考叶绿体基因组NC_049022。而对菝葜属三物种(S.china、S.glabra、S. glyciphylla)的突变热点区域的分析则使用DNAsp(v5.10)[43],窗口长度600bp,步长100bp。
1.5 编码区域进化分析
通过对 3 个菝葜属物种(S. china、S.glabra、S.glyciphylla)所共有的76 个基因两两之间Ka/Ks 的计算[44],以衡量不同基因上所受的选择压力。随后对84个物种中rbcL基因进行两两比对,寻找受正选择压力的物种。
1.6 简单重复序列分析
使用MISA 软件[45-46]对菝葜的全叶绿体基因组序列中所包含的SSR 位点进行开发,使用的参数为单核苷酸至少重复8 次,二核苷酸与三核苷酸至少重复4次,四、五以及六核苷酸至少重复3次。
SSR 引物的设计在 NCBI 完成(https://www.ncbi.nlm.nih.gov/tools/primer-blast),前后引物长度为18-22bp,GC含量40%-60%,前后引物距离SSR位点30bp以上,最终产物长度在100 到500bp。PCR 体系总量25μl ,其中 DNA 1μl,前引物 1μl,后引物 1μl ,ddH2O 9.5μl,2×EsTaqMaster Mix 12.5μl。PCR 程序设置为95 °C 前处理 3min,后在 95°C 处 30s、50°C 处 30s、72°C处 30s 进行扩增并循环 35 次,最后在 72°C 处 10 min 进行延伸。本实验选取表1中的3个菝葜样品DNA作为所有引物扩增的模板,在扩增完成后取6μl 最终的PCR 产物进行琼脂糖凝胶电泳,检测是否为单一的清晰条带且片段长度是否与预测结果相符,最后通过Sanger法测序进行评估。
2 结果
2.1 菝葜叶绿体基因组的结构特征与基因成分
如表1 所示,采用不同策略开展组装所得叶绿体基因组结果存在明显差异。其中下载自NCBI 的参考序列 NC_049022 中的ycf3、ycf4及psbN基因分别被注释为pafI、pafII及pbf1,Nanopore 测序数据独立组装结果中基因顺序发生了整体颠倒,且其中IR区域有部分基因出现3 次复制。而Illumina 测序数据独立组装结果的LSC 区域中trnL-UAA和trnF-GAA基因未出现。结果表明基于Illumina 与Nanopore 数据整合组装得到的菝葜叶绿体基因组结果则最为准确,比NCBI 参考序列短584bp,但能精确注释86 个编码基因。如图1所示,菝葜叶绿体基因组组装结果最终呈现典型的四分结构。如图2所示,混合组装具有明显优势,更能体现现实中菝葜叶绿体基因组序列的真实情况。
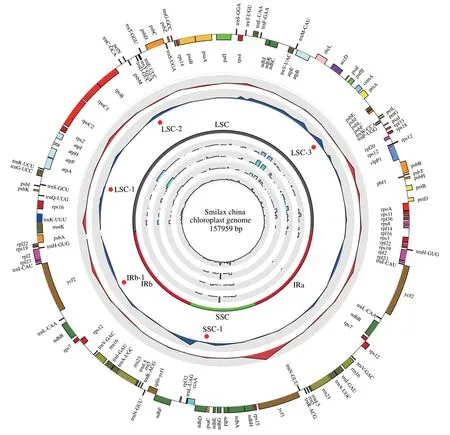
图1 基于Unicycler混合组装的菝葜叶绿体基因组

图2 差异较大区域的序列比对验证
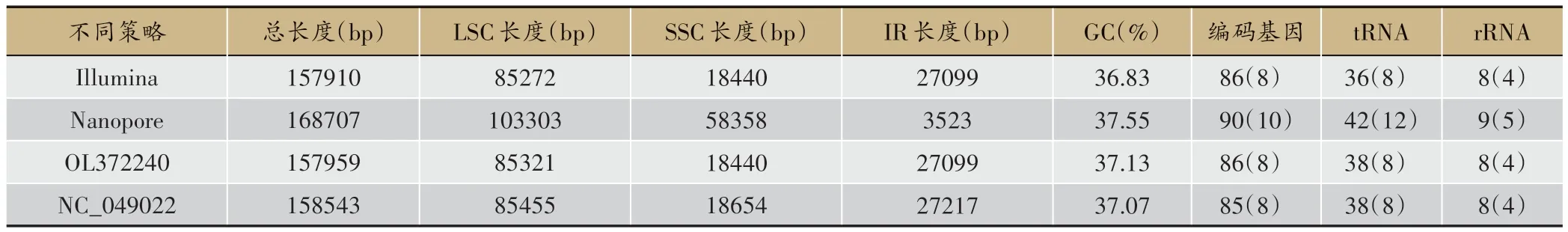
表2 不同测序技术所得菝葜叶绿体基因组特征
注:定位在trnT-UGU-trnL-UAA之间的 A(71 bp)、B(8 bp)和C(20 bp)与trnL-UAA中的D(44 bp)和E(15 bp)与petA-psbJ之间的F(32 bp)以及rpl33-rps18之间的H(9 bp),均显示Sanger 测序结果与OL372240更加一致;定位在rpl33-rps18之间的 G(34 bp)和 I(5 bp),显示Sanger测序结果与NC_049022更加一致。
2.2 系统发育分析
如图3 所示,百合科被分为两个大支,菝葜族的3个物种聚为一支,并与百合族与油点草族共同为聚为一个较大的支,最终与重楼族、藜芦族与嘉兰族得到百合科主要的两大支之一,而另外一个大支由黄精族、沿阶草族、铃兰族、龙血树族、天门冬族、萱草族、丝兰族、吊兰族、葱族以及芦荟族组成;其中菝葜属中约26.5Mya开始分化,百合族约83.7Mya出现分化。
2.3 叶绿体基因组的比较分析
本研究表明菝葜及其近缘物种的IR 区域为最保守的区域,但IR 区域的收缩与舒展仍应为常见,最终可导致叶绿体基因组大小的改变。将菝葜属的5个物种IR 边界与其近缘物种进行比较(图3A),用以考察IR 区域的收缩与扩张。IRb 区域与LSC 区域的分界线在T.altaica里位于rps19基因中,而在除S.glabra的菝葜属物种则出现了一定的扩张,到达了rpl22基因内;与此同时,菝葜与萱草中的SSC 区域的序列顺序与其他物种的方向完全相反。
如图3B 所示,基于mVISTA 对七个叶绿体基因组的分析表明,其中五个菝葜属物种序列相似度较高,与同科不同属物种差异主要集中在SSC 区域的ndhF到ndhD之间,而在LSC 区域则较为普遍,主要位于非编码区域之中。

图3 菝葜与其近缘物种的比较分析
另外,利用DNAsp 鉴别菝葜属3 物种(S.china、S.glabra、S.glyciphylla)全叶绿体基因组的突变热点区域(图1 第八环)。核苷酸多态性值范围为0 到0.05278,其中ndhH区域的核苷酸多态性最高,为0.05278,随后是ndhE-psaC区域、rps15-ndhH区域以及pafII-cemA区域,其中除pafII-cemA区域处于LSC 区域外另三个均位于SSC区域中。
2.4 编码区进化研究
本研究使用同义置换率(Ks)与非同义置换率(Ka)来决定特定基因上可能存在的选择压力。如图1第五、六、七环所示,通过计算S. china、S. glabra、S.glyciphylla等三个物种所共有的76个蛋白质编码基因的Ka/Ks值以确定这些基因上所受的选择压力。通常认为Ka/Ks 大于1 则认为该基因受到了正选择,小于1则受到纯化选择,等于1 时则可能出现中性进化[47]。计算表明,在S. china和S. glyciphylla之间有accD与rbcL的Ka/Ks分别为1.304和1.597,S.china与S.glabra之间有ndhE为 1.721,而S.glabra与S.glyciphylla之间同样为ndhE其Ka/Ks 为1.724。认为上述几个基因可能受到环境的正选择压力。对84 个物种间rbcL基因的比对分析认为,有部分物种间存在正选择信号(图4)。

图4 基于9个基因联合数据集构建的百合科植物系统发育树及84物种间rbcL基因的正选择情况
2.5 SSR位点搜索与引物筛选
通过对菝葜叶绿体基因组中的SSR 位点的统计,总共得到了247 个SSR(单核苷酸152 个,二核苷酸70个,三核苷酸12 个,四、五核苷酸各6 个、六核苷酸1个)。菝葜叶绿体基因组中有102 个SSR 位点处于基因之中,另外145 个则位于基因间区之中(图1 第二,三环)。如图5所示,利用琼脂糖凝胶电泳法对设计的5 对引物进行筛选,选取上述的3 个菝葜样本作为模板。其中LSC-1、SSC-1、IRb-1 位点来自叶绿体基因组的不同区域,均有明亮的扩增结果,同时Sanger 测序结果表明它们的引物扩增得到的产物长度为298bp、242bp 及268bp,存在序列长度差异,具备作为分子标记的潜力。

图5 验证5对SSR引物的琼脂糖凝胶电泳图
3 讨论
随着技术的进步与成本的降低叶绿体基因组的研究已十分普遍,但受手段限制NCBI 上原有的叶绿体基因组序列在准确性上可能存在一定的问题,对于下载自NCBI 的数据可以通过叶绿体基因组所存在的DNA 条形码,如rbcL、matK以及psbA-trnH等片段来对其正确性进行考察,是一种快速准确的鉴别手段[48-49],可以通过中药材DNA条形码鉴定系统进行验证(www.tcmbarcode.cn)。
本文共运用三种策略对菝葜叶绿体基因组进行了有参组装(reference-based assemble),联立Sanger 法测序的验证结果,表明利用混合组装技术获得的序列信息(OL372240)具有明显优势,更能体现现实中菝葜叶绿体基因组的真实情况。如表1 所示,长短读长混合组装菝葜叶绿体基因组能够进行更加精确的注释,与其相比短读长数据独立组装结果中出现了1452 bp的空隙;长读长数据独立组装结果中存在部分基因错误复制的现象。如图2 所示,在trnT-UGU-trnL-UAA(A、B 与 C)、trnL-UAA(D 和 E)、petA-psbJ(F)以 及rpl33-rps18(H)的区间,OL372240 与 Sanger 测序结果高 度 一 致 ;在rpl33-rps18(G 和 I)的 区 间 ,原 NC_049022 测序结果可能更准确。短读长组装叶绿体基因组虽然具有很高的准确性,但采用限制性内切酶构建短片段测序库时,可能遗留部分缺乏酶切位点的序列使得测序结果存在缺失[50]。然而,新近研究则认为独立的长读长Nanopore 测序结果足以拥有极高的精度[51],不过更多研究则倾向于利用短读长和长读长混合组装叶绿体基因组[16,52]。

表3 本实验设计的5对SSR引物
关于对菝葜及其近缘物种系统发育关系确认的研究中通常使用不同物种所共有基因或全叶绿体基因组作为数据集[26,53-54],这里依据OrthoFinder软件得到全部物种的叶绿体基因组编码基因序列中的直系同源基因建立了数据集。此外,在中国植物志分类中同属于百合科萱草族的玉簪属(Hosta)和萱草属(Hemerocallis)在本文中所得到的系统发育树中处于两个不同的分支之中,但在与其他文献系统发育树的比对中并未发现明显的拓扑结构差异。在对分化时间的研究中,菝葜属的5 个物种以较高的后验概率(P= 100%)聚为一支,其中白背牛尾菜首先分化出来,其分歧时间为26.52Mya,处于渐新世夏特期,随后菝葜与另一物种于17.17 Mya 分化,位于中新世布尔迪加尔期,认为上述两个时期的环境存在使菝葜属物种出现分化的因素存在,有待其他菝葜属物种的进一步验证。
目前叶绿体基因组DNA 条形码研究表明,全叶绿体基因组或多位点作为超级条形码可以弥补单位点DNA 条形码在近缘物种鉴别上的固有限制[11]。在本研究中核苷酸多态性分析认为菝葜叶绿体基因组突变热点部位主要包括ndhH、ndhE-psaC、rps15-ndhH以及pafII-cemA等区域,而选择压力分析表明rbcL、ndhE及accD基因在菝葜属3 物种中受到正选择压力,此外菝葜属的SSC区域在与同科其他物种的比对中也展现了较大的整体差异性,它们均有作为物种鉴别DNA条形码的潜力。同时,在本研究中出现正选择的3 个基因中rbcL基因主要与光合作用与光呼吸作用相关,accD基因主要与脂肪酸的合成有关[55],而ndhE基因则与NADH 脱氢酶相关,其中rbcL基因在很多科植物中受到普遍的正选择压力[56],认为可能是环境的变化导致菝葜属物种光合作用的改变。此外,在菝葜属内叶绿体基因组结构近似,但与其他近缘属物种相比明显的IR区域的扩张现象可以显示其进化事件的存在,另外,土茯苓(S. glabra)出现了与其他菝葜属不同的现象,且在其IR 与SSC 区域存在ycf1作为假基因的情况,值得进一步考察。关于pafI、pafII以及pbf1基因的注释一直存在一定的争议,在过往的其他叶绿体基因组中也存在同样的情况[57-58],而其实际注释方式有待深入研究。
本研究最后开发了 LSC-1、SSC-1、IRb-1 等来自于叶绿体基因组的不同区域的SSR 标记位点,未来可以作为分子标记的补充,用于菝葜及其近缘属种中药材资源的分类鉴定,而如图5 中的LSC-2 和LSC-3 条带较为暗淡可能是由于菝葜叶绿体基因组中目标SSR序列出现了变异导致。LSC-3测序结果表明其序列中存在明显的差异,表明该位点在不同地区的菝葜中存在一定的差异,有进一步开发潜力。
