反复发作的遗传性胰腺炎1例及其家系突变分析
2022-08-23任晓侠葛库库刘欢宇张含花韩亚楠王华车凤玉方莹
任晓侠 葛库库 刘欢宇 张含花 韩亚楠 王华 车凤玉 方莹
西安市儿童医院消化科,西安 710003
【提要】 遗传性胰腺炎是一种罕见的常染色体显性遗传疾病,约占胰腺炎总数的8.7%,常在儿童期发病,表现为反复发作的急性胰腺炎。该病增加了慢性胰腺炎和胰腺癌的患病风险。现报道1例14岁男性青少年慢性胰腺炎的诊治经过,经二代测序技术最终证实为SPINK1及CFTR双基因杂合变异所致遗传性胰腺炎。
遗传性胰腺炎是一种罕见的常染色体显性遗传疾病,约占胰腺炎总数的8.7%[1],常在儿童期发病,表现为反复发作的急性胰腺炎,该病增加了CP和胰腺癌的患病风险,在1952年由Comfort和Steinberg发现并确立[2]。对于遗传性胰腺炎的系统性研究报道多见于欧洲和北美洲,日本也有相关报道[2-4],而中国报道则较少,且多数是以个例报道和综述为主,没有系统的大规模研究。遗传性胰腺炎好发于儿童和青少年,起病年龄多在20岁前,并且母系遗传患儿的发病年龄(约在9岁)早于遗传自父亲的患儿(约在14岁)[5]。临床表现及遗传学特点复杂多样,主要特征为与急性胰腺炎症状类似的反复腹痛,但其发病年龄较早,且呈家族聚集性。现有研究认为,引起该病的变异基因主要包括PRSS1、SPINK1、CTRC、CPA1、CFTR、CEL/CEL-HYB、CLDN2、TRPV6、CaSR、AP2S1等[6-8]。本文报道1例随访4年的SPINK1及CFTR双基因变异的难治性反复发作的青少年慢性胰腺炎,对其发病特征、疾病管理及基因突变情况进行探讨。
一、临床资料
患者男,14岁。因“反复上腹部疼痛2年”入院。2016年7月(11岁)发病,进食高脂食物后出现剧烈上腹痛,能耐受,伴恶心呕吐,当地查血淀粉酶806 U/L,以“急性胰腺炎”收入当地医院,给予禁饮食、抑酸、抑酶、补液等治疗12 d后症状好转出院。其后类似症状反复发作6次,均住院非手术治疗后症状缓解出院。既往无吸烟饮酒史,无外伤手术史,否认家族遗传病史。入院体检:发育正常,营养中等,神志清,精神欠佳,无黄染、皮疹,心肺未见异常。腹平坦,触软,上腹压痛,无反跳痛、肌紧张,未触及包块,肝脾未触及,Murphy征阴性,无移动性浊音,肠鸣音5次/min。实验室检查:血常规、凝血功能及肝功能正常,血钙2.33 mmol/L,血淀粉酶74 U/L,尿淀粉酶261 U/L,葡萄糖3.8 mmol/L。2018年6月7日腹部CT示胰腺未见异常;MRCP示腹、背侧胰管未见汇合,考虑胰管先天发育异常,胰腺分裂,胰管轻度扩张,肝内外胆管未见异常。入院后给予禁饮食、抑酶、抑酸、补液治疗。2018年7月3日在全麻下行ERCP,术中见主乳头无异常,副乳头略肿大,开口无异常。经主乳头插管成功,0.035导丝进入主胰管体尾部,造影可见主胰管全程显影,最宽处内径约2 mm,无明显狭窄,无异常透光区,可见背侧胰管显影,内径约2 mm,吸引可见造影剂、胰液排出通畅,退镜终止检查。ERCP诊断:胰管造影未见异常。术后患儿无腹痛、发热、出血等不适,血常规、血淀粉酶未见异常。腹痛缓解出院,嘱出院后忌暴饮暴食、避免高脂饮食等诱发因素。建议基因检测,家长拒绝。后患儿病情反复,多次按急性胰腺炎住院行内科治疗。2019年2月复查腹部CT平扫示胰腺多发钙化灶(图1A、1B);MRCP示胰腺信号不均匀,胰管扩张(图1C),肝内外胆管未见异常,考虑诊断为CP。
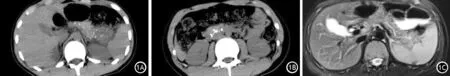
图1 患儿腹部影像学征象。腹部CT平扫示胰腺多发钙化灶(1A、1B); MRCP示胰腺信号不均匀,胰管扩张(1C)
二、家系调查
患儿父亲及母亲非近亲婚配,无任何胰腺炎病史及临床症状;患儿舅舅家表弟9岁时有复发性胰腺炎病史,共发作5次,控制饮食后未再发病(图2)。否认家系中成员遗传性疾病病史。

注:Ⅱ-1为患儿父亲,Ⅱ-2为患儿母亲,Ⅲ-2为患儿,Ⅲ-4为患儿表弟图2 遗传性胰腺炎患儿家族谱系图
三、基因检测
为查明病因,在获得其父母的书面知情同意后,对患儿及父母进行了基因检测。基因序列分析结果显示,患儿7号染色体CFTR基因的10外显子上的NM_000492:c.1351G>A(p.Gly451Arg)杂合变异(图3),5号染色体SPINK1基因4号外显子上的NM_003122:.93_101delATGTTACAA(p.31_33del)LysCysTyr杂合变异(图4)。患儿母亲携带上述两个杂合变异,患儿父亲未携带此变异,基因检测结果见表1。对患儿表弟及其父母进行基因测序,均未发现携带相关变异基因。
四、病情演变及随访情况
2020年3月13日患儿再次突发腹痛,急查血淀粉酶1 288 U/L,血脂肪酶1 062 U/L,给予生长抑素抑酶、艾司奥美拉唑抑酸、禁饮食等治疗,酶学恢复正常。3月20日行ERCP+EPT+胰管支架置入术,术中见主胰管全程扩张,副胰管显影、扩张,最宽约6 mm,行数字减影血管造影(digital subtraction angiography,DSA)可见胰管内较多透光区(图5A),胰管括约肌切开,球囊清理出大量蛋白石(图5B)。循导丝置入7Fr-9 cm、7Fr-7 cm单猪尾胰管支架于主胰管内(图5C、5D)。术中留置鼻空肠营养管,行半要素饮食喂养,经口少量流食。术后随访1年,患儿腹痛缓解,临床未再反复,复查血淀粉酶、脂肪酶无升高。4个月后拔除空肠营养管,6个月后一枚支架自行脱落,1年后内镜下拔除剩余另一枚支架,经口低脂饮食,体重增长15 kg,复查胰腺影像学较前无加重。2018年随访至2022年3月,患儿全程监测临床症状体征、酶学、影像学改变等,通过临床干预、疾病指导,自2020年3月ERCP术后至2022年3月,2年间临床症状未再反复。目前16岁6月,身高178 cm,体重63 kg,体重指数为19.8 kg/m2。

图3 遗传性胰腺炎患儿(3A)、患儿父亲(3B)、患儿母亲(3C)CFTR基因分析结果

图4 遗传性胰腺炎患儿(4A)、患儿父亲(4B)、患儿母亲(4C)SPINK1基因分析结果

表1 患儿基因检测相关结果

图5 患儿ERCP术中图像。DSA示胰管内较多透光区(5A);球囊清理出大量蛋白石(5B);DSA示双枚单猪尾胰管支架于主胰管内(5C);双枚单猪尾胰管支架内镜图像(5D)
讨论本例患儿3年余14次住院,排除其他胰腺炎致病因素,通过基因检测发现其携带SPINK1基因c.93-101delATGTTACAA(p.31-33delKCY)杂合变异及CFTR基因c.1351G>A(p.G451R)杂合变异,并且母亲携带这两个杂合变异,可以合理地得出结论,遗传性胰腺炎是该青少年反复发作急性胰腺炎的重要病因。
Sanger测序家系验证显示患儿母亲携带上述两个杂合变异,患儿父亲未携带。由于遗传性胰腺炎存在不完全外显,因此其母亲并未表现相关症状。本例患儿携带的CFTR基因c.1351G>A(p.G451R)杂合变异,为CP相关的已知变异。Zou等[9]对1 061例中国汉族CP患者进行靶向测序发现1例患者携带该杂合变异,同时在1 196名正常对照人群中未发现该变异;该变异未见正常人群数据库报道;生物信息学蛋白功能预测软件SIFT、PolyPhen_2、MutationTaster预测均为有害。根据美国医学遗传学与基因组学学会(ACMG)指南,该变异定义为临床意义未明。患儿携带的SPINK1基因c.93_101delATGTTACAA(p.31_33delKCY)杂合变异同样为CP相关的已知变异。Qian等[10]在1例16岁患有CP的患者中检测到该变异,在正常人群数据库中的频率是0.00001,为低频变异;生物信息学蛋白功能预测软件MutationTaster预测为多态性。根据ACMG指南,该变异定义为临床意义未明。
SPINKl基因突变导致抑制胰蛋白酶活性水平下降,从而引起对胰腺炎的易感性增加[11]。SPINKl基因常见的突变位点包括N34S、P55S(多见于欧洲和美国)以及IVS3+2TC(常见于亚洲)[12],该基因的突变可能会使胰腺炎的发病风险增加23%[13]。CFTR是囊肿性纤维化遗传病的致病基因,有研究发现50%以上CFTR基因突变的遗传性胰腺炎患者年龄超过30岁[14],约有1.5%的囊肿性纤维化患者会患数次胰腺炎。如果患者同时携带多个突变基因,那么其胰腺炎发病率会显著增加[15]。 本例患儿同时携带2个基因变异,因此其反复发病频率高。
目前针对遗传性胰腺炎患者尚无特异性的治疗方法,治疗原则与其他原因导致的胰腺炎相同[16]。北美儿科胃肠病学、肝病学和营养学会胰腺委员会强调解决CP患儿的疼痛管理[17],同时建议在需要内镜引流时采用EUS。ERCP及胰管结石引流可使CP患儿症状得到改善,减少胰腺炎发作[18-19]。结合本例患儿胰腺影像学动态变化,胰腺逐渐出现胰管扩张及多发钙化灶,行ERCP乳头括约肌切开、胰管取石及胰管支架置入后,患者症状缓解,1年内未再出现胰腺炎复发。
由此可见,对于临床上反复发作胰腺炎的儿童和青少年,可通过基因测序寻找潜在的致病因素,从基因检测中获得的信息有利于进行家庭筛查以及发现其他异常,也可用于指导患者的长期治疗。遗传性胰腺炎具体发病机制不明,且不同的致病基因所存在的突变位点也尚未被完全发现,其拷贝数与遗传性胰腺炎的确切关系也未明确,因此实现遗传性胰腺炎快速基因诊断及个性的基因治疗仍需要进行长期、大量的研究。
利益冲突所有作者声明无利益冲突
