一种面向癌症个性化的代谢分析方法
2022-08-18冯昌奇郑浩然
冯昌奇 郑浩然
0 引言
肿瘤异质性是癌症治疗的主要障碍之一,它可以发生在多个层面,包括基因、分子和组织学等[1-3]。由于基因的改变,癌细胞之间在疾病过程中出现了表型和功能上的差异[4]。同样,肿瘤细胞与其微环境的相互作用或血管生成和缺氧的局部变化在肿瘤中也是不均匀的。这种巨大的生物、细胞和组织异质性存在于瘤内水平(一个肿瘤内的分子差异)、患者内水平(一个患者病变间的肿瘤特征差异)和患者间水平(患者间肿瘤特征的差异)。这种异质性影响了肿瘤的侵袭性和耐药性,是有效治疗策略设计的一个重大挑战[5]。
细胞代谢在癌症[6]和其他各种疾病[7]的发病机制中起着重要的作用。癌细胞重新规划其能量代谢,以满足增殖和迁移的能量需求。侵袭和转移的机制是复杂的,死亡率主要是由癌症进展到转移状态[8]引起的。了解正常细胞和癌细胞之间的代谢差异可以阐明疾病的发展机制,也可能有助于确定治疗性代谢靶点。
在系统生物学领域,基因组尺度代谢模型(genome-scale metabolic model,GEM)整合了可用的组学数据和基因组序列,以提供对有机体细胞内代谢的更好的机制理解。将转录组学数据整合到基因组规模的代谢网络中,以执行网络级别的模拟,这是一个有用的步骤,可以将数据规范化,并试图从转录组中表达的酶的综合证据推断代谢状态。GEM可以作为一种强大的工具来对患者进行分层,或确定癌症代谢中的关键中介基因,使生物学途径相互交织,以及提供癌细胞能量和生物合成需求的代谢物[9-10]。此外,GEM还可用于识别新的靶点和药物,并在药物发现或药物重新定位的背景下创建和/或测试假设[11-12]。
目前已有大量的研究关注癌症代谢上的异质性。例如,Berndt等[13]应用系统生物学方法对10例人类肝细胞癌(hepatocellular carcinoma,HCC)的代谢变化进行了分析,发现最大的肿瘤间异质性是乳酸的摄取和释放以及细胞糖原含量的大小。Baloni等[14]对来自癌症基因组图谱(The Cancer Genome Atlas,TCGA)的1 156个乳腺正常和肿瘤样本的基因表达数据进行了散度分析,构建了个性化的特异性代谢网络,根据它们的不同代谢情况将样本划分成了4个类群,并识别出了两种最具抗肿瘤活性的药物。但目前有关肿瘤代谢异质性的研究大都关注部分通路的代谢变化或肿瘤代谢系统的异常代谢,很少关注肿瘤细胞在整个代谢系统上发生的代谢变化之间的异质性。
为了探究不同癌症患者在代谢系统改变上的异质性,本文基于679例患者的癌细胞和正常细胞的基因表达数据,构造了679个个性化的环境特异性代谢网络模型。通过对这些模型进行研究,课题组希望分析不同个体在代谢变化上的独特性和普遍性,以便根据需要设计药物治疗方案。
1 数据与方法
1.1 数据源
本文从基因组数据共享(GDC)门户网站(https://gdc.cancer.gov/)下载了TCGA中癌症样本的RNA-seq数据,其中包含了来自33种癌症类型的60 483个基因的预处理基因表达数据。本文将研究对象定在其中的17种癌症类型上,因为它们包含了足够数量的正常细胞样本,利于比较癌症细胞与原发组织细胞之间的代谢差异。为了方便进行后续的差异分析,本文使用TCGA数据库level3层面中的read count数据。
TCGA的每个样本都有自己的编号作为唯一标识,其中包含了项目名称、组织来源、参与者编号和样本类型等信息。为了对不同的患者进行个性化的分析,本文根据参与者编号对样本进行匹配,筛选出同时具有癌症细胞和正常细胞基因表达数据的样本。最终,本文筛选出了679对样本,样本数的详细分布见表1。

表1 样本在17种癌症类型中的数量分布Table 1 The number distribution of samples in 17 cancer types
1.2 差异表达分析
差异表达分析以具有生物学意义的方式计算基因表达,然后通过对基因表达量进行统计学的分析,找到具有显著差异的基因。在本文的分析中,对每一对来自同一患者的癌症和正常细胞样本,使用Limma[15-16]R包对其进行差异表达分析,以log2FC > 1.5和P值<0.01作为显著差异的阈值,筛选出在癌症细胞中表达水平显著上调的基因。这些基因代表了癌细胞相对其原发组织显著活跃的部分。
1.3 生成环境特异性代谢网络
在本研究中,课题组使用当前最新的人类基因组规模代谢网络模型Recon3D[17]进行癌症患者的个性化环境特异性代谢网络模型的生成。Recon3D中包含了2 248个代谢基因和10 600个代谢反应,并且提供了基因-蛋白-反应(gene-protein-reaction,GPR)关联用于整合基因和反应之间的关系。
在这一步中,本文首先从差异表达分析中获得的差异上调基因中筛选出Recon3D中所包含的基因,之后结合GPR将这些基因映射到代谢反应上,作为生成特异性代谢网络模型的核心反应集合,代表了癌症细胞相对其原发组织显著活跃的那部分反应。特别地,为了保证生成的特异性代谢网络能够进行正常的生物质合成,本文中将生物质合成反应添加到核心反应集合中。
之后,本文使用COBRA Toolbox v3.0[18]实现的FASTCORE[19]算法,基于Recon3D生成环境特异性代谢网络模型。根据FASTCORE算法的特性,生成的模型能够在包含核心反应集合的条件下,满足最小通量一致性。生成的代谢网络模型代表了特定患者的癌细胞相对其正常细胞发生异变的那部分代谢系统,不同模型之间的差异充分体现出了癌症患者之间代谢异常的独特性。
1.4 鉴别特异性代谢网络的潜在靶点
在本研究中,潜在靶点的鉴别分为两步:基因敲除和毒性测试。
在基因敲除阶段,本课题组使用RAVEN[20]工具箱提供的removeGenes函数模拟基因缺失后的网络。对特异性网络中的基因,每次模拟删除其中的一个,然后计算敲除后模型的生物质合成速率。如果一个基因被敲除后导致模型的生物质合成速率变成0或几乎为0,那么认为这个基因对它所在的特异性网络模型来说是必要的,其缺失会导致模型不能合成生长所需要的物质,进而无法维持生存。
将一个基因作为潜在的靶点,除了要满足其缺失会影响到特异性网络生存的条件外,还应该对正常细胞是无害的。所以,在毒性测试阶段,本文从代谢图谱(metabolic atlas)[21]下载了83个正常细胞的代谢网络模型,在这些网络上模拟必要基因的缺失。通过检查缺失后的模型能否完成包括能量与氧化还原、内转化、底物利用和产物生物合成等在内的56个代谢任务[22]来确定该癌症必要基因能否作为潜在的治疗靶点。
2 结果
2.1 差异表达基因
本文中对每个成对样本分别进行了差异表达分析,以获得特定患者的癌细胞中显著上调的基因。679例患者的差异表达基因数量分布如图1(a)所示。可以看出,不同患者癌细胞相对正常细胞的差异表达基因数量具有很大的差异,无论是上调还是下调。经过课题组的统计,发现在多数样本的结果中,差异下调基因的数量多于上调基因,这可能意味着癌细胞普遍会更多地抑制正常细胞基因的表达。特别地,没有任何一个基因出现在全部样本的上调基因或下调基因集合中,并且平均每对样本有2.5个下调基因和1.5个上调基因是独一无二的,不在其他任何样本的差异表达基因中出现,这些信息充分体现了癌症在不同患者体内发生改变的独特性,这种独特性是研究普适性治疗方案的主要障碍,有必要得到生物医学研究人员的重视。
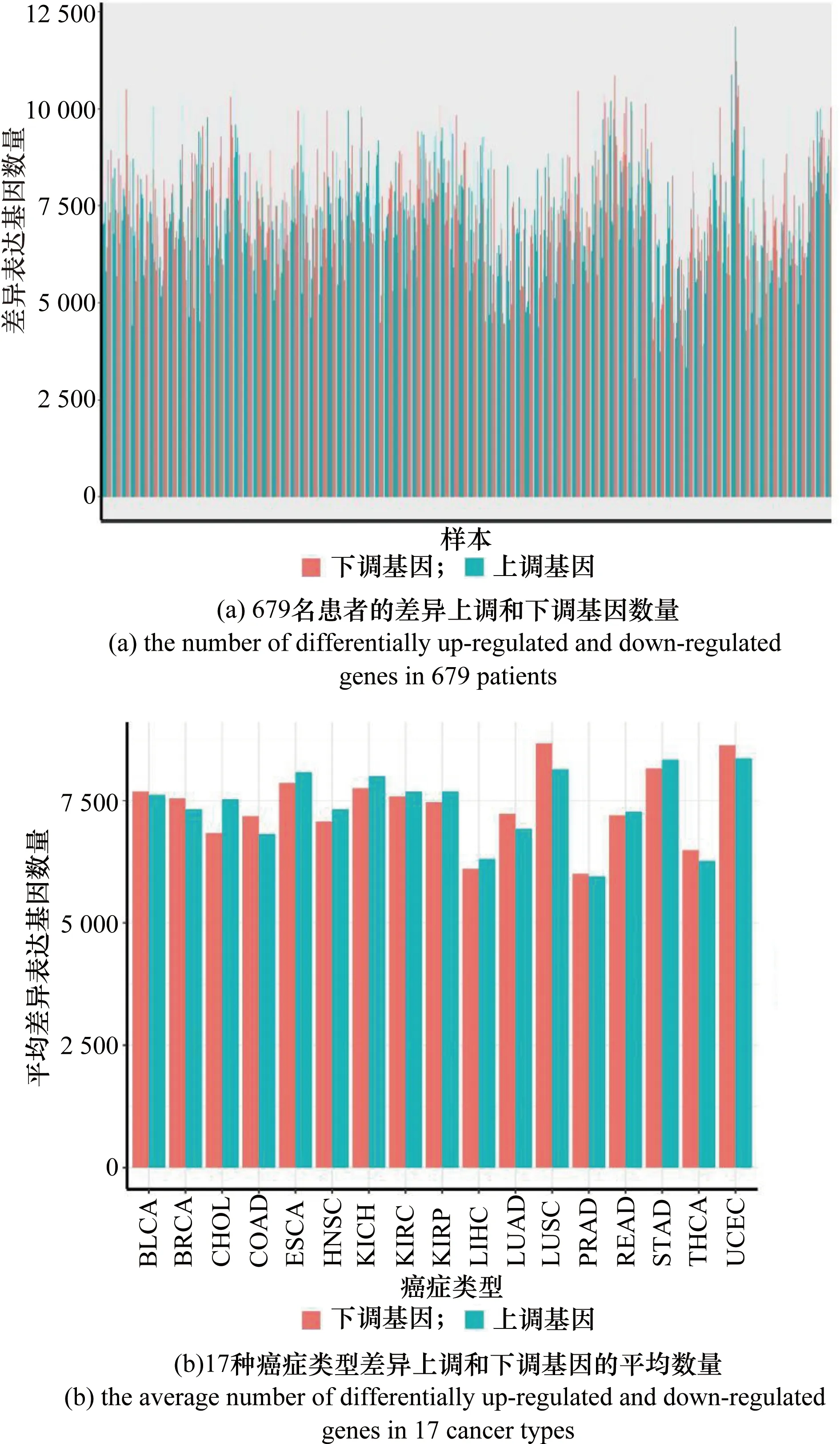
图1 差异表达基因数量Figure 1 Number of differentially expressed genes
为了更好地分析不同癌症类型之间在差异表达基因上的不同,本文计算了每种癌症类型所有患者的平均差异表达基因数量,结果如图1(b)所示。可以看出,LIHC、PRAD、THCA 3种类型的差异表达基因数量明显比其他癌症要少,这意味着这3种癌症相对其他癌症来说,发生改变的程度更小。而相反的,LUSC、STAD和UCEC发生的变化更加明显。
2.2 个性化特异性代谢网络模型
本文分别将679对样本进行差异表达分析得到的差异上调基因与人类基因组规模代谢网络模型Recon3D结合,获取了679个个性化的环境特异性代谢网络模型,这些模型充分反映了每位患者癌细胞相对其原发组织细胞发生变化的代谢系统。679个代谢网络模型的基因和反应规模如图2所示。
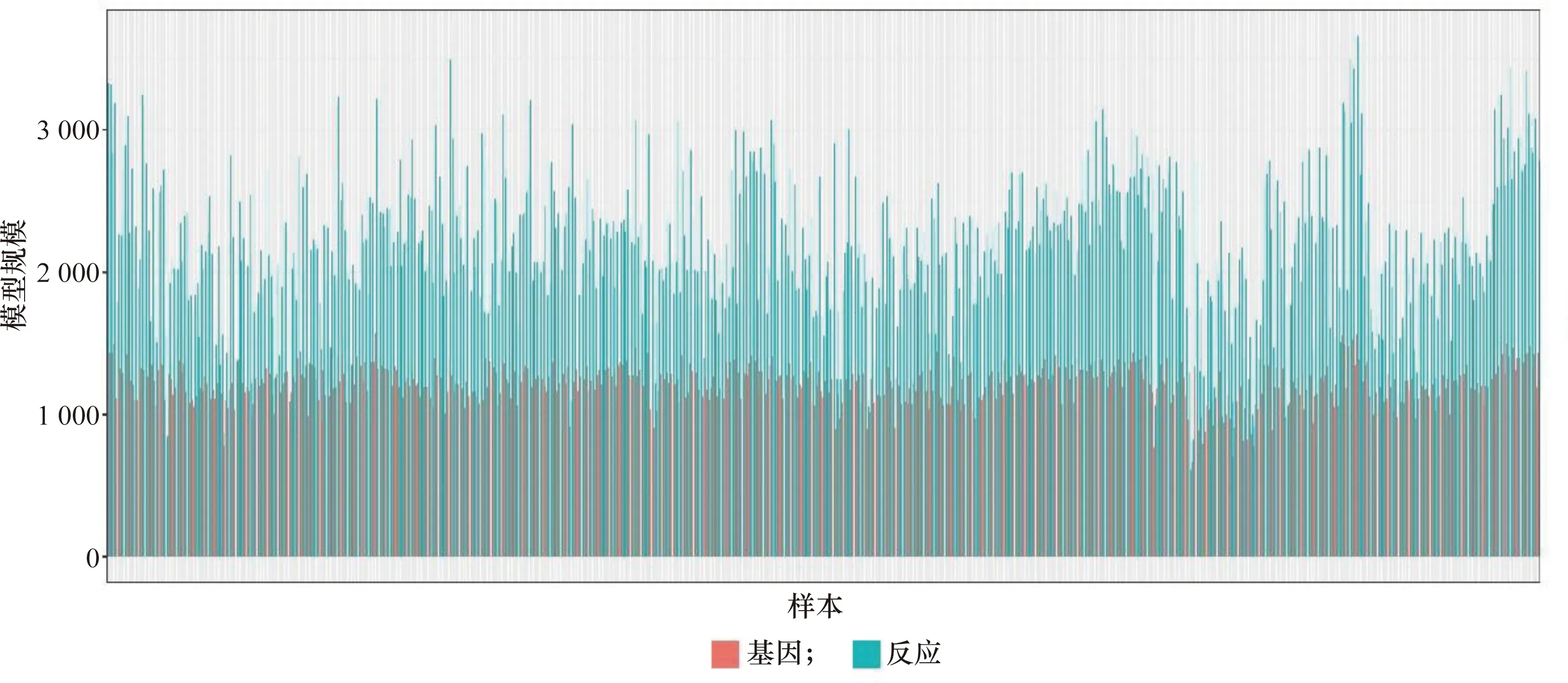
图2 679个个性化代谢网络的规模Figure 2 The scale of 679 personalized metabolic networks
相比于包含2 248个代谢基因和10 600个代谢反应的Recon3D而言,个性化的特异性代谢网络平均只包含1 217个代谢基因和2 181个代谢反应,在去除了多余信息的同时,充分反映了不同患者癌细胞发生改变的代谢系统的独特性。
本文统计了每种癌症类型的个性化代谢网络模型的平均规模,发现LIHC、PRAD和THCA 3种类型的网络平均规模最小,这与差异表达基因得到的结果是一致的,即这3种癌症的癌细胞相对正常细胞发生的代谢变化程度最小。
2.3 潜在靶点基因
从代谢网络模型中删除一个基因可能会对整个代谢系统的生长产生巨大的干扰,也可能几乎没有影响。为了分析代谢系统中可能引发干扰的基因,本文对679个个性化代谢网络模型进行了基因缺失的测试。如果一个基因被敲除后会影响其所在模型的生长,那么认为这个基因是该模型的必要基因。如果同时该基因的缺失又不会影响到正常组织细胞的正常代谢活动,即认为这个必要基因的缺失对正常细胞是无影响的,可以作为候选的治疗靶点。
本文对679个模型分别进行了基因敲除和毒性测试,并计算了每种癌症类型下的模型平均必要基因和潜在靶点数量,如图3所示。特别地,PRAD的平均必要基因和潜在靶点数量显著高于其他癌症类型。结合前面差异表达基因和代谢网络模型的结果,本课题组考虑这是因为PRAD相对其他癌症来说,发生变化的那部分代谢网络规模更小,因而没有更多的同工酶和同工反应来保证生物质的合成,因此其网络结构在稳定性上较差。
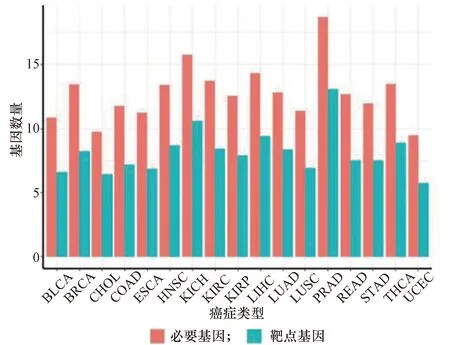
图3 17种癌症类型的平均必要基因和靶点数量Figure 3 The average numbers of necessary genes and targets for 17 cancer types
本文统计了679个模型的潜在靶点,得到227个不重复的基因,这些基因的缺失会影响一个到若干个模型中生物质的合成速率。227个靶点基因在679个模型中出现次数的分布如图4所示。可以看出,有93%(211/227)的基因在少于100个模型中被认为是潜在的靶点,其中绝大多数(195/211)出现在679个模型中的出现次数少于30次。这意味着对一例患者可行的治疗靶点对其他患者大概率是无效的,从而导致大多数的药物治疗方案可能只能对极少数的患者具有治疗意义。
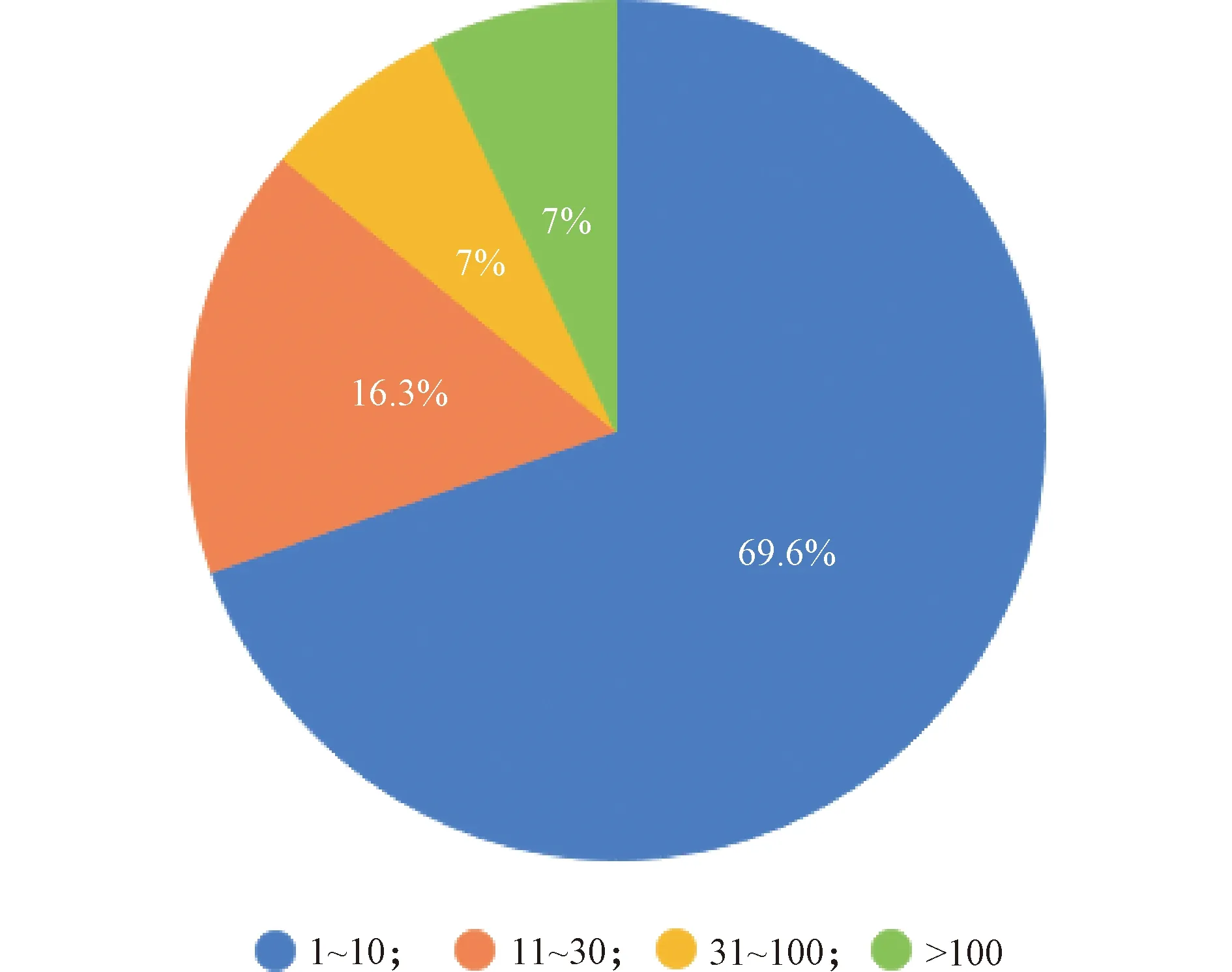
图4 227个靶点基因在679个模型中出现次数的分布Figure 4 Distribution of the occurrence number of the 227 target genes in 679 models
特别地,69.6%(158/227)的基因只在少于10个模型中被认为是潜在的靶点,基于这些基因设计的药物治疗方案将具有极高的特异性。其中,又有63个基因的缺失只在某一个特定的模型中影响生物质的合成,这些基因被认为是针对特定患者的特异性治疗靶点。这些特异性治疗靶点中的大多数已经被各项研究确定为可能的癌症药物靶点或被证实与癌症的发生、进展和转移有关联。例如,B3GTN3在胰腺癌[23]和宫颈癌[24]的进展和转移中发挥了显著的作用,与不良预后相关,被预测为这两种癌症的治疗靶点。IDH2的突变在神经胶质瘤中被发现,但这种突变并不常见,研究者们分析了87例神经胶质瘤病例,只有1例发生了IDH2突变[25],间接体现了IDH2在癌症治疗中的特异性。ME1的过表达与乳腺癌肿瘤体积大、生存期差和化疗耐药性成正相关,被预测为乳腺癌潜在的预后标志物和治疗靶点[26]。有研究利用miR-885-5p抑制了胃癌中ME1的过表达,有效地抑制了癌细胞的增殖和发展,改善了预后不良的状况[27]。NNMT的表达在卵巢癌[28]和胃癌[29]中显著升高,并与术后预后不良相关,被认为是卵巢癌治疗的重要靶点[30]。PHGDH在乳腺癌和胰腺癌细胞的增殖、迁移和侵袭中发挥调节作用,可能是乳腺癌和胰腺癌重要的预后指标和治疗靶点[31-32]。有研究通过抑制剂下调PHGDH的表达,显著抑制了膀胱癌细胞的增殖能力并诱导细胞的凋亡[33]。
63个特异性靶点基因中大部分已经被先前的研究证实可以作为癌症的潜在靶点或与癌症密切相关,没有被证实的部分则代表了日后进一步开展癌症个性化分析和治疗的潜在研究对象。有趣的是,本文中发现有14个特异性靶点都属于溶质运载蛋白家族,这可能意味着这一家族的基因在癌症进展中发挥了重要作用,其研究价值有待深入发掘。
3 讨论与结论
在本文中,课题组使用来自TCGA的17种癌症类型的679例患者的癌症细胞样本和正常组织细胞样本,分析了不同癌症患者的癌细胞相对正常组织细胞在代谢表型和代谢网络层面上发生的变化。本文的结果表明,无论是在代谢表型上还是在更深层的代谢网络上,不同的癌症患者细胞癌变后发生的代谢异常都在很大程度上是不同的。在代谢表型上,不同患者的差异表达基因有很大的差异,且平均每位患者有2.5个下调基因和1.5个上调基因是独有的,且没有任何一个基因在所有患者的差异表达基因中出现。在代谢网络层面上,不同患者癌细胞发生改变的代谢系统规模也不尽相同,这反映了发展癌症个性化治疗的必要性。
本文的实验结果发现了若干个性化的靶点基因,这些基因中的一部分已经被证实与癌症相关,另一部分则为生物医学研究提供了新的线索。但由于目前的实验分析都停留在理论层面,缺少相应的临床医学实验验证,因此本研究仍然具有一定的局限性,得到的结果对于临床肿瘤个性化治疗的应用价值还需要进一步的研究。在未来的工作中,课题组将尝试扩展可用的样本数量和覆盖的癌症类型,以期不断提高实验结果的准确性和可信度。
