Generation and Characterization of Monoclonal Antibodies Against Red-Spotted Grouper Nervous Necrosis Virus (RGNNV)
2022-08-17QINYinghuiLIUJiaoyunLUYuananandLIUXueqin
QIN Yinghui,LIU Jiaoyun, LU Yuanan, and LIU Xueqin, *
Generation and Characterization of Monoclonal Antibodies Against Red-Spotted Grouper Nervous Necrosis Virus (RGNNV)
QIN Yinghui1), 2), 3),LIU Jiaoyun1), 2), 3), LU Yuanan4), and LIU Xueqin1), 2), 3), *
1),,430070,2),430070,3),,430070,4),,
Nervous necrosis virus (NNV) can infect more than 120 fish species worldwide and has caused high mortality and sig- nificant economic losses to the aquaculture industry. Among different genotypes of NNV, the red-grouper nervous necrosis virus(RGNNV) is the most widely distributed one with the highest number of susceptible fish species.In this study, the capsid protein (Cp) gene of RGNNV was recombined and expressed instrain BL21 (DE3) and the recombinant Cp (rCp) was used as an immunogen to produce monoclonal antibodies (MAbs) through hybridoma cell fusion technology. Three MAbs were produced and characterized by indirect enzyme-linked immunosorbent assay (ELISA), western blotting, and immunofluorescence assay (IFA). Wes- tern blotting result showed that the MAbs could specifically react with the capsid protein of RGNNV. The result of IFA showed that the MAbs could recognize virions in RGNNV-infected grouper spleen (GS) cells. These results indicate that the MAbs can specifi- cally recognize RGNNV virions and can be used to produce a rapid detection method. This study provides a foundation for further studies on the rapid diagnosis of RGNNV and its infection mechanisms.
RGNNV; monoclonal antibody; capsid protein; grouper
1 Introduction
Nervous necrosis virus (NNV) is an icosahedral, non- enveloped, positive-sense, single-stranded RNA virus be-longing to thegenus of the Nodaviridae family (Doan., 2017). NNV was first isolated from diseased striped jack larvae, which was designated as striped jack nervous necrosis virus (SJNNV), and identified as a new member of thefamily in 1992 (Mori., 1992). To date, NNV has been detected from at least 120 fish species in marine and freshwater environments through- out the world, with the notable exception of South Ameri- ca (Bandín and Souto, 2020). In severe outbreaks, NNV infection could cause mass mortality up to 100% in larvae and juveniles, resulting in a massive economic loss world- wide (Bandín and Souto, 2020).
The genome of NNV is bipartite, comprising of two po- sitive-sense RNA molecules (RNA1 and RNA2) (Doan., 2017). The RNA1 encodes an approximate 110kDa RNA- dependent RNA polymerase (RdRp), also named protein A, which is responsible for viral genome replication (Guo., 2004). The RNA2 encodes an about 42kDa capsid protein (Cp), which is the sole structural protein of NNV, and an ideal target for RGNNV detection and an ideal al- ternative for the generation of MAbs against RGNNV (Chen., 2015). Based on the phylogenetic analysis of a small variable sequence of RNA2, namely the T4 region, NNV has been traditionally classified into four genotypes: red- spotted grouper nervous necrosisvirus (RGNNV), striped jack nervous necrosis virus (SJNNV), tigerpuffer nervous necrosis virus (TPNNV) andbarfin flounder nervousne- crosis virus (BFNNV) (Doan., 2017; Bandín and Sou- to, 2020). Among them, the genotype of RGNNV is the most widely distributed one with the highest number of susceptible host species (Doan., 2017; Bandín and Souto, 2020).
RGNNV vaccines containing the capsid protein as an- tigen include subunit vaccine (Lin., 2018), virus-like particle vaccine (Chien., 2018), DNA vaccine (Vale- ro., 2016), inactivated vaccine (Pakingking., 2010)and live vaccine (Nishizawa., 2012). Through these vaccines which are effective for protecting grouper from RGNNV infection in the experiment, commercial vaccines or efficient antiviral drugs against RGNNV is still the ma- jor difficulty for the control of this viral disease.At pre- sent, early diagnosis of viral infection and timely imple- mentation of measures are still the ideal methods for con- trolling RGNNV. Therefore, it is necessary to develop ra- pid and convenient detection methods and to elucidate the infection mechanism of RGNNV. Immunoassays based on specific monoclonal antibodies (MAbs) are more suited to the field detection of viral infections due to their sensiti- vity, specificity, low cost, speediness, and ease of proce- dures (Tang., 2018, Hassantabar, 2021). MAbs can also be employed as a tool for identifying virus target organs and investigating host-virus interactions and infec- tion mechanisms (Tang., 2018, Zhong., 2019). Therefore, specific MAbs against RGNNV are urgently needed.
In the present study, the Cp of RGNNV was cloned and expressed, and was used as the immunogen for MAbs pro- duction. The produced MAbs were characterized by indi- rect enzyme-linked immunosorbent assay (ELISA), immu- nofluorescence assay (IFA), and western blotting. All re- sults indicate that the three MAbs can recognize the coat protein of RGNNV and can be used to detect RGNNV in infected GS cells.
2 Materials and Methods
2.1 Ethics Statement
All experimental animals were used in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols for animal care and handling used in the present study were approved by the InstitutionalAnimal Care and Use Committee of Huazhong Agricul-ture University (HZAU). To minimize animal suffering, all experimental mouses were anesthetized with 3-Aminoben- zoic acid ethyl ester methanesulfonate (MS-222) (Sigma, USA) before sacrificing and handling.
2.2 Cell and Virus
Grouper fin cells (GF-1) were provided by Prof. Li Lin (Chen, 2017) and grouper spleen (GS) cells were provided by Prof. Qiwei Qin (Huang., 2015). GF-1 and GS cells were cultured in L-15 medium (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and maintained at 25℃. RGNNV isolated from adult Hulong grouper in 2017 and preserved in our laboratory was used in this study (Qin., 2020). The virus was replicated in GF-1 cells grown in L15 with antibiotics and 2% FBS at 25℃. The supernatant was harvested upon the appearance of extensive cytopathic effect (CPE), and then stored at −80℃ until used.
2.3 Cloning, Expression, and Purification of Cp
Based on the genome sequence of the Cp of RGNNV(GenBank no. MN309752), the specific primers (Cp-F: 5’-CGATGGTACGCAAAGGTGAGAAG-3’, Cp-R:5’-CCTCGTTTTCCGAGTCAACCCTGG-3’) were designed to amplify the target gene encoding the Cp. Total RNA was extracted from infected GF-1 cells using Trizol reagent (Takara, Japan) following the manufacturer’s instructions. The quality and quantity of total RNA were checked using a NanoDrop ND-8000 spectrophotometer (Thermo Scientific). cDNA was synthesized from 1μg of RNA using the commercial PrimeScript™ RT Reagent Kit (Takara, Japan) according to the manufacturer’s instruct- tions. The resulting cDNA was used to amplify the Cp gene and The PCR product of the Cp gene was purified and di- gested with the restriction enzymes EcoRI and XhoI, and then ligated into pET-28a plasmid to construct pET-28a- Cp recombinant plasmid, which then was transformed intoBL21 (DE3). The positive clone was confirmed by sequencing,BL21 with positive clone were incu- bated in LB medium to a midlogarithmic phase and induced by adding 0.5mmolL−1isopropyl β-D-1-thiogalactosidase (IPTG). The rCp was purified by His Trap™ HP Ni-Aga- rose (GE Healthcare) following the manufacturer’s ins- tructions. The induced bacteria lysate and purified rCp were analyzed by SDS-PAGE and visualized after stain- ing with Coomassie brilliant blue R-250. The concentra- tion of rCp was determined using the Bradford method(Bradford, 1976).
2.4 Mouse Immunization and Monoclonal Antibody Production
Four BALB/c mice were immunized intraperitoneally (i.p.) with 100μL of 1mgmL−1purified rCp emulsified with an equivalent volume of Freunds complete adjuvant (FCA, Sigma). After two weeks, a similar injection was adminis- trated using Freund’s incomplete adjuvant (Sigma) instead of FCA. Booster immunization were then given twicethe tail vein at one-week intervals. Three days after the last immunization, the mice were sacrificed. Spleen cells were collected from the immunized mice and fused with mye- loma cells (SP2/0) using 50% polyethylene glycol 4000 (Sigma). The cells were distributed into 96-well culture plates (Costar) in 1640 medium (Hyclone) supplemented with 1% HAT (Gibco), and the culture medium was changedevery 3–5d. After 10–12d, the supernatant fluids from those wells growing hybridomas were screened by indirect ELISA. Hybridomas with positive results were subcloned three times by the limiting dilution method until all hy- bridoma subclones secreted monoclonal antibodies reac- tive to rCp. Then the MAbs were characterized by indi- rect ELISA, western blotting, and IFA.
2.5 Indirect Enzyme-Linked Immunosorbent Assay (ELISA)
The specificity of MAbs was detected by indirect ELISA as described by Tang. (2018). Briefly, 100μL purified rCp (2μgmL−1) was coated into each well of 96-well mi- croplates (Costar) at 4℃ overnight. Then the wells were washed three times with phosphate buffered saline (PBS) containing 0.05% Tween-20 (PBST) and later blocked with 200μL of 5% bovine serum albumin (BSA) in PBS for 1h at 37℃. After washing three times with PBST buffer, the wells were incubated with 100μL hybridoma culture super- natant as a primary antibody for 1h at 37℃. After wash- ing, 100μL goat-anti-mouse Ig-alkaline phosphatase con- jugated (Abclonal, China) diluted 1:4000 in PBS was add- ed as a second antibody and incubated for 1h at 37℃. Following a final wash, 100μL 0.1% (w/v) p-nitrophenyl phosphate (pNPP, Sigma, USA) in 50mmolL−1carbonate-bicarbonate buffer (pH 9.8) containing 0.5mmolL−1MgCl2was added to each well and incubated at room tempera- ture for 10min in the dark. The reaction was stopped by adding 50μL of 2molL−1NaOH to each well and absor- bance was measured with an automatic ELISA reader at405nm. As a negative control, the myeloma culture super- natant instead of the MAbs culture supernatant was added as the first antibody. Each experiment was repeated in tri- plicate.
2.6 Preparation of Mouse Ascites
After screened by ELISA, western blotting, and IFA, the three positive hybridomas were cultured with 1640 me- dium in 24-well plates at 37℃ supplied with 5% CO2se- parately. Then, 106hybridomas were collected into a 1.5mL centrifuge tube and washed with a 1mL preheated 1640 medium at 37℃, followed by centrifuging at 1000for 5min and resuspending with 500μL 1640 medium. The hy- bridomas were intraperitoneally injected into the eight-week-old BALB/c mice, which had been given an injection of 500μL liquid paraffin one week ago. About 10 days after injection, the mice were anesthetized with the ether, and the ascites with anti-Cp MAbs was collected with a 10mL syringe, placed at 4℃ overnight and centrifuged at 760for 10min. After removing the lipids in the upper layer, the ascites was centrifuged at 12000for 30min again. Final- ly, the supernatant was collected and further purified by the caprylic acid-ammonium sulfate precipitation method(Zhong., 2018), and then completely dialyzed in PBS at 4℃ and stored at −80℃ for further analyses.
2.7 Virus Purification
The method of virus purification was is according to the previous report (Qin, 2020) with some modifications. Briefly, 200mL of the supernatant of the infected SSN-1 cells was centrifuged to remove cellular debris at 10000for 1h and the recovered supernantant was filtered througha 0.22μmolL−1filter. The filtrate was centrifuged at 150000for 1h at 4℃. The recovered pellet was resuspended in 10mmolL−1phosphate buffered saline (PBS, pH 7.4) and centrifuged again at 150000for 1h at 4℃ through a semi-discontinuous 20%-35%-50% (W/V) sucrose gradient in PBS (10mmolL−1, pH 7.4). The virus was collected from the interface between 35% and 50% sucrose and pelleted bycentrifugation at 150000for 40min. The pelleted viral particles were resuspended in PBS (10mmolL−1, pH 7.4) and the purity was checked by electron microscopy.
2.8 Western Blotting Assay
The western blotting assay was used to analyze the spe- cificity of MAbs. GS cells were infected by RGNNV for 36h; then GS cells were lysed to collect the lysates. Samples were separated by 10% SDS-PAGE and transferred to a PVDF membrane (Merck Millipore, Darmstadt, Germany).The membrane was blocked overnight with 5% BSA in TBST buffer at 4℃, and then incubated with MAbs (di- luted 1:2000) at 37℃ for 1h. After washing the membrane three times with TBST buffer, the membrane was incubated with goat-anti-mouse Ig-horseradish peroxidase (Abclonal, China) as the secondary antibody at 37℃ for 1h. After washing three times, the membrane was visualized by en- hanced chemiluminescence reagents.
2.9 Indirect Immunofluorescence Assay (IFA)
GS cells were seeded onto cover slips according to the method described by Tang. (2018). Briefly, acid-etch-ed circular cover slips were kept in 24-well plates, and 105cellswell−1were seeded and incubated to allow cells to attach to the cover slips. After 24h incubation, the me- dium was removed and the wells were carefully washed with PBS. Cells were then infected with RGNNV at two multiplicity of infection (MOI) and incubated at 25℃ for 24h post-infection. Monolayers were then rinsed with PBSand fixed with cold acetone for 10min. After washing with PBS, the cells were blocked overnight with 5% BSA in PBST buffer at 4℃. Then the cells were incubated with MAbs at 37℃ for 1h. The myeloma culture supernatant instead of the MAbs was added as the negative control. After washing three times with PBST, the slides were in- cubated for 45min at 37℃ in the dark with goat anti-mouseIg-FITC (1:200, Abclonal), and 4,6-diamidino-2-phenyl- indole (DAPI, Roche) staining (blue) was used to visual- ize cell nuclei. Slides were rinsed again and then mounted with 50% glycerin and observed under a fluorescence mi- croscope (Olympus DP70).
2.10 Statistical Analysis
All data management, analysis, and graphing were con- ducted using GraphPad Prism 7.0 (GraphPad Software, Inc.). The significance of the variability between different treat- ment groups was determined by one-way ANOVA. The dif- ference was considered significant when<0.05.
3 Results
3.1 Expression and Purification of the Cp
SDS-PAGE revealed that the Cp of RGNNV was suc- cessfully expressed inBL21 (DE3) with the pET- 28a system after IPTG induction. The distinct band about 43kDa (Fig.1, lane 2) was in accordance with the predict- tion of a 37kDa Cp plus a His-tagged protein of 6kDa. After purification with the Ni2+affinity chromatography, rCp with high purity was obtained (Fig.1, lane 5).
3.2 Production, Selection and Subtype Identification of MAbs
A total of 56 hybridomas were observed under an in- verted microscope and 16 of these hybridomas gave posi- tive results compared with myeloma culture supernatant by indirect ELISA. Among the positive cells, three hybrido- mas with higher absorbance were finally cloned and sub- cloned by limiting dilution (Fig.2A). The negative control using the myeloma culture supernatant showed a very low background (O.D.<0.1).
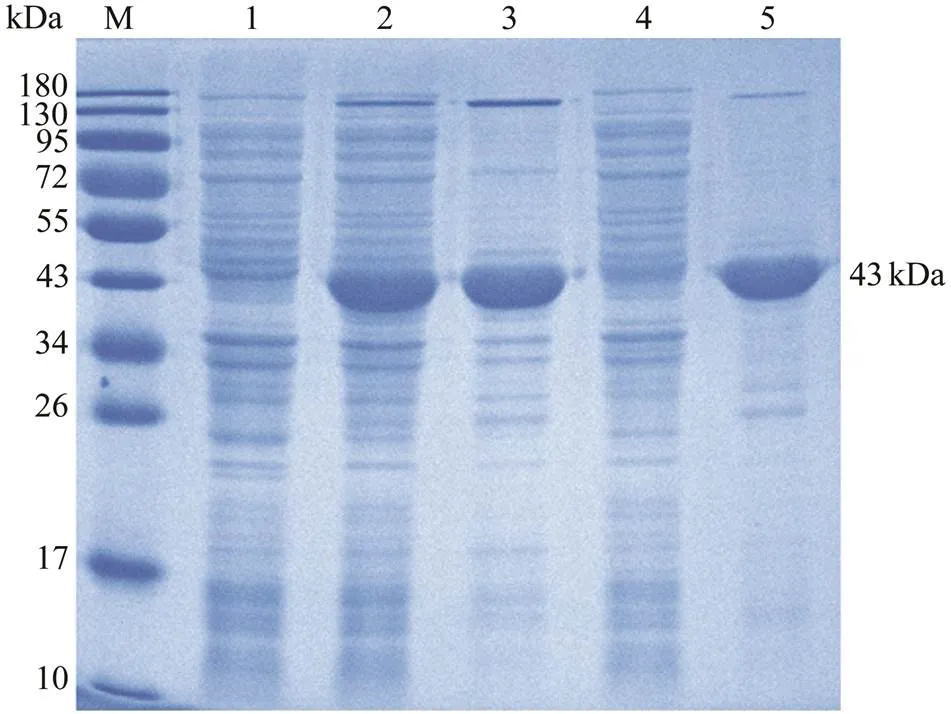
Fig.1 Expression, purification, and SDS-PAGE analysis of recombinant capsid protein (rCp). M, marker; Lane 1, transformed E. coli without IPTG induction; Lane 2, trans- formed E. coli induced with IPTG; Lane 3, the precipitateof transformed E. coli induced with IPTG after sonication; Lane 4, the supernatant of transformed E. coli induced with IPTG after sonication; Lane 5, purified rCp.
To identify the subtype of the three MAbs, purified as- cites of 1C5, 2B2 and 3D6 were subjected to SDS-PAGE, transferred to a PVDF membrane, and then incubated with HRP-conjugated goat-anti-mouse immunoglobin (Ig). Re- sults showed that the three MAbs were all IgG-type with a heavy chain about 50kDa and a light chain about 25kDa (Figs.2B, C).
3.3 Virus Purification
High-purity RGNNV virus particles were achieved by sucrose density gradient centrifugation. The size of the vi- rion was about 30nm. It was non-enveloped and icosahe- dral (Fig.3A). SDS-PAGE results showed purified RGNNV has a single protein band about 37kDa (Fig.3B), which was in accordance with the fact that coat protein was the only structure protein RGNNV and its molecular weight was 37kDa.
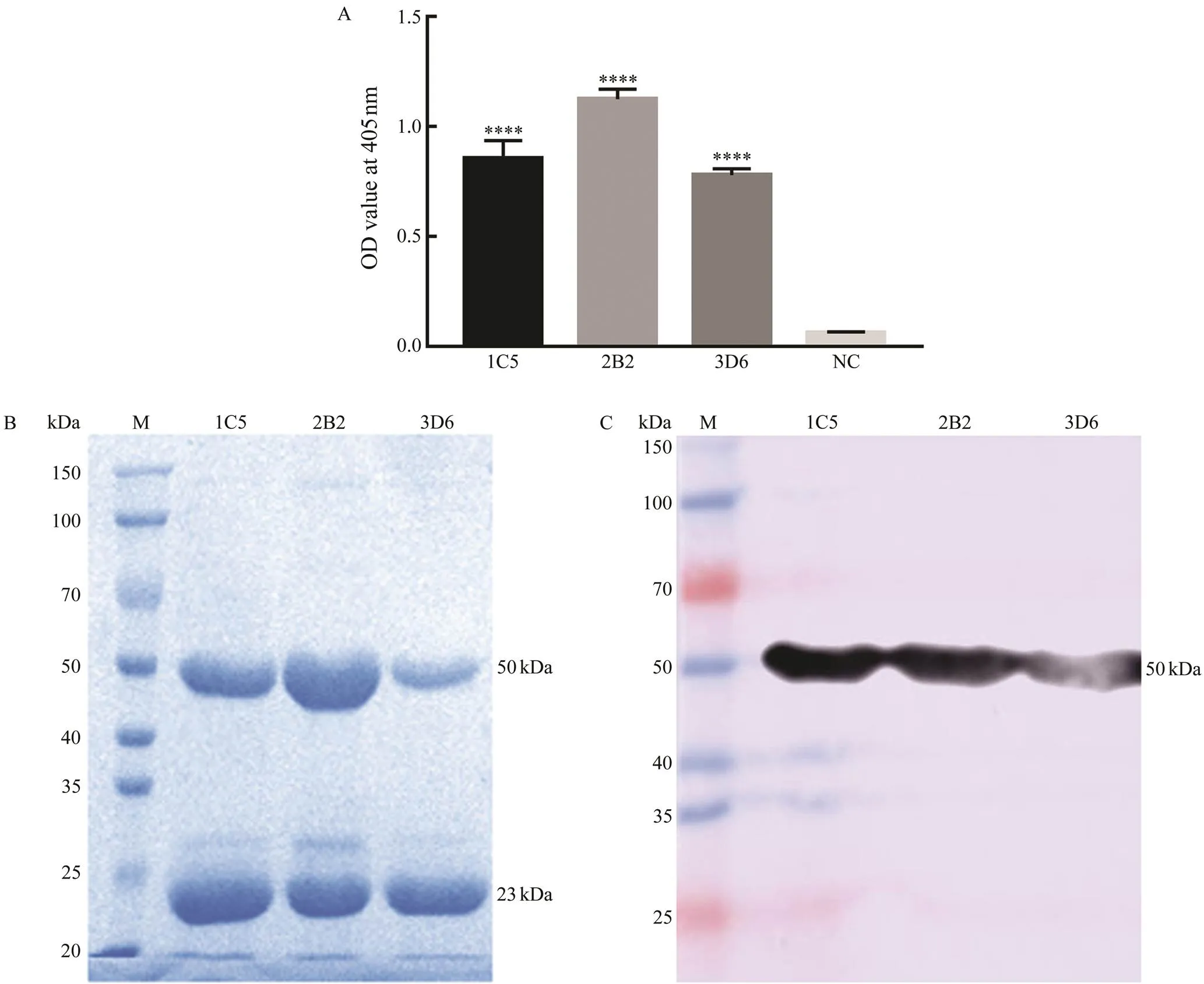
Fig.2 Screening and subtype identification of anti-Cp monoclonal antibodies (MAbs). (A) The three anti-Cp MAbs reacted with the rCp by ELISA. The culture myeloma culture supernatant instead of primary antibody was used as a negative control. Error bars represent standard deviations obtained by measuring each sample in triplicate. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. (B) SDS-PAGE analysis of the purified three anti-rCp MAbs. The bands show the 50kDa heavy chain and 25kDa light chain of IgG. (C) The three anti-rCp MAbs were identified as IgG subtype by western blotting.
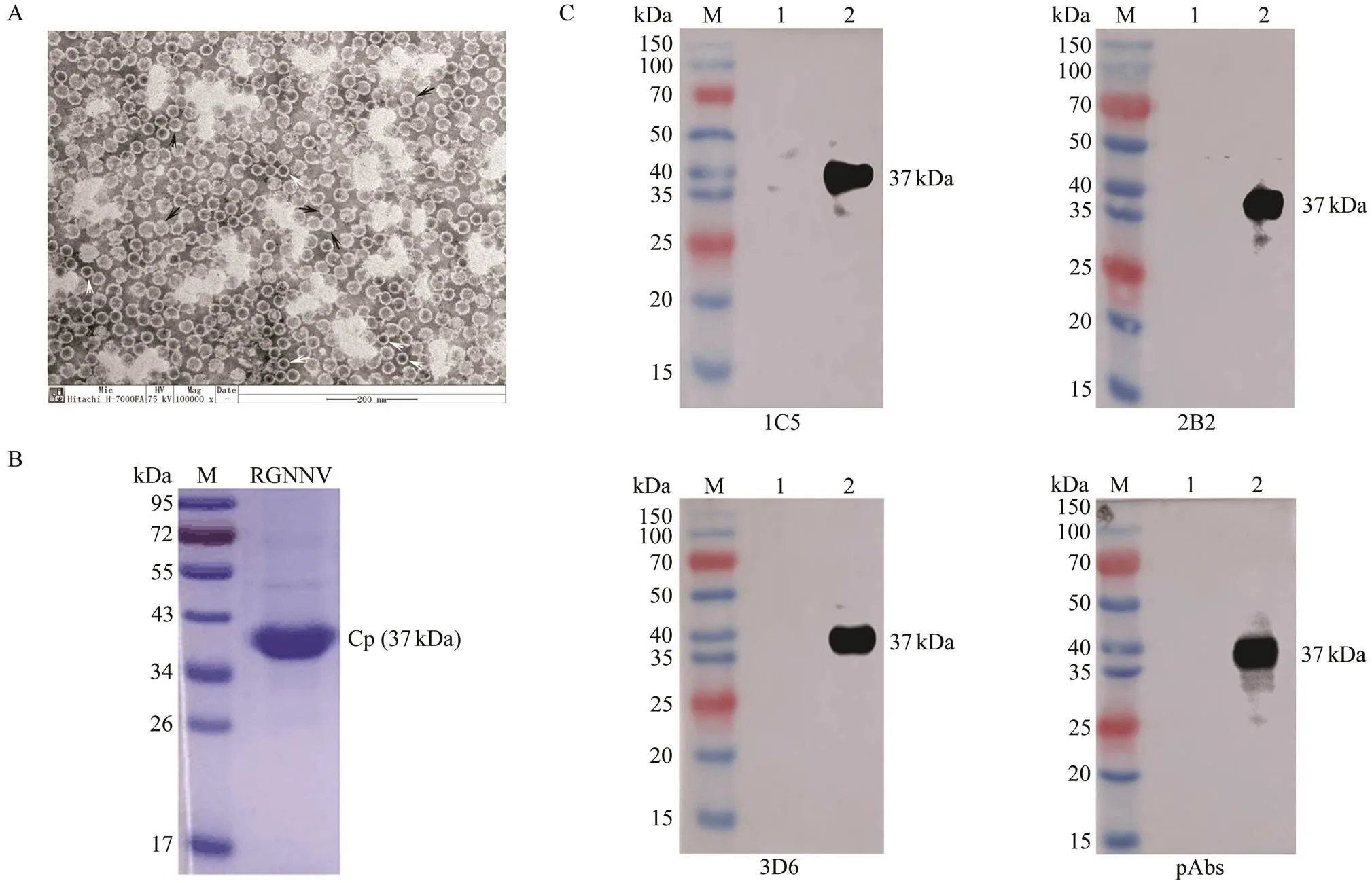
Fig.3 Virus purification and western blotting analysis of monoclonal antibodies (MAbs) with RGNNV-infected GS cells. (A) Electron micrograph of negatively stained virus particles pelleted from infected GF-1 cells culture fluid, showing ico- sahedral virus particles with a diameter about 30nm. Black arrowheads indicate complete virions, white arrowheads indicate empty virions. Bar=200nm. (B) SDS-PAGE of purified RGNNV. One specific protein band with molecular masses about 37kDa was detected by electrophoresis. Lane M, molecular mass marker; Lane RGNNV, purified RGNNV. (C) Western blotting analysis of monoclonal antibodies (MAbs) with RGNNV-infected GS cells. Lane M, molecular mass marker; Lane 1, uninfected GS cells; Lane 2, RGNNV-infected GS cells. 1C5, MAbs 1C5; 2B2, MAbs 2B2; 3D6, MAs 3D6; pAbs,anti-rCp polyclonal antibodies.
3.4 Western Blotting
The results of western blotting analysis showed that the three MAbs could recognize an about 37kDa protein inRGNNV-infected GS cells (Fig.3C), whose molecular mass was consistent with RGNNV Cp (Fig.3B). In contrast, no positive band was found when the samples of SDS-PAGE were replaced by uninfected GS cells.
3.5 Indirect Immunofluorescence Analysis (IFA)
To identify whether the MAbs could specifically recog- nize RGNNV, the specific combination of RGNNV-in- fected GS cells and MAbs was analyzed with IFA. All three antibodies showed strong green fluorescence signals in theRGNNV-infected GS cells. In contrast, no positive sig- nals were observed in the uninfected cells (Fig.4). These results demonstrate that the three MAbs could specifically recognize RGNNV in RGNNV-infected cells and could be further used to detect RGNNV in diseased fish.
4 Discussion
NNV infects more than 120 fish species worldwide and causes high mortality and significant economic losses tothe aquaculture industry. Among different genotypes of NNV,the RGNNV genotype is the most widely reported with the highest number of susceptible species (Bandín and Souto, 2020). The high pathogenicity, together with the nonavail- ability of commercial vaccines or drugs against RGNNV, highlights the urgent need to develop simple and rapid diag- nostic methods. Among multiple diagnostic methods, im- munoassays based on MAbs are more suited to the field detection of viral infections (Wang and Zhan, 2006). There- fore, it is significant to produce specific MAbs against RGNNV for the development of rapid diagnosis methods and investigation of infection mechanisms of RGNNV. However, traditional MAbs production methods require abundant high-purity virus particles, which are extremely difficult to obtain (Tang., 2018). Here, therefore, the high-purity rCp of RGNNV was used as the immunogen for MAbs production, and three MAbs specific to RGNNV were obtained.
ELISA showed that the three MAbs could react with rCp. To further ensure whether the MAbs could specifically re- cognize the coat protein of RGNNV, the high-purity RGNNV virus particles were achieved by sucrose density gradient centrifugation (Fig.3A). The virions were small (about 30nm), non-enveloped and icosahedral with features similar to betanodaviruses (Chen, 2015; Xing, 2020). SDS-PAGE showed the high-purity RGNNV has a speci- fic protein band about 37kDa, whose molecular mass wasin accordance with the coat protein of RGNNV (Chen, 2015). Moreover, the 37kDa protein in RGNNV-infected GS cells could be recognized by the MAbs in a westernblotting assay, which showed the MAbs could recognize the coat protein of RGNNV specifically. IFA showed that the three MAbs could recognize RGNNV in RGNNV-in- fected cells and could be further used to detect RGNNV in diseased fish (Fig.4). Moreover, IFA staining observa- tions of infected GS cells exhibited two patterns on the lo- calization of Cp, one with nucleolus and cytoplasmic stain- ing in 25% of GGNNV-infected GS cells and the other with- out nucleolus labeling. This result was well supported by previous studies, which showed that N-terminal amino acids 23–31 of RGNNV Cp is a nuclear export signal and Cp is located in both cytoplasm and nucleolus in a part of trans- fected and infected cells (Guo, 2003). Moreover, Qin. (2020) observed that abundant RGNNV particles and bodies were presented on the cytoplasm of RGNNV-infect- ed GS cells by transmission electron microscopy. There- fore, our results demonstrated that the developed MAbs can react with the recombinant coat protein and the native coat protein of RGNNV, suggesting that the recognized epitopes were linear and conformation-independent.
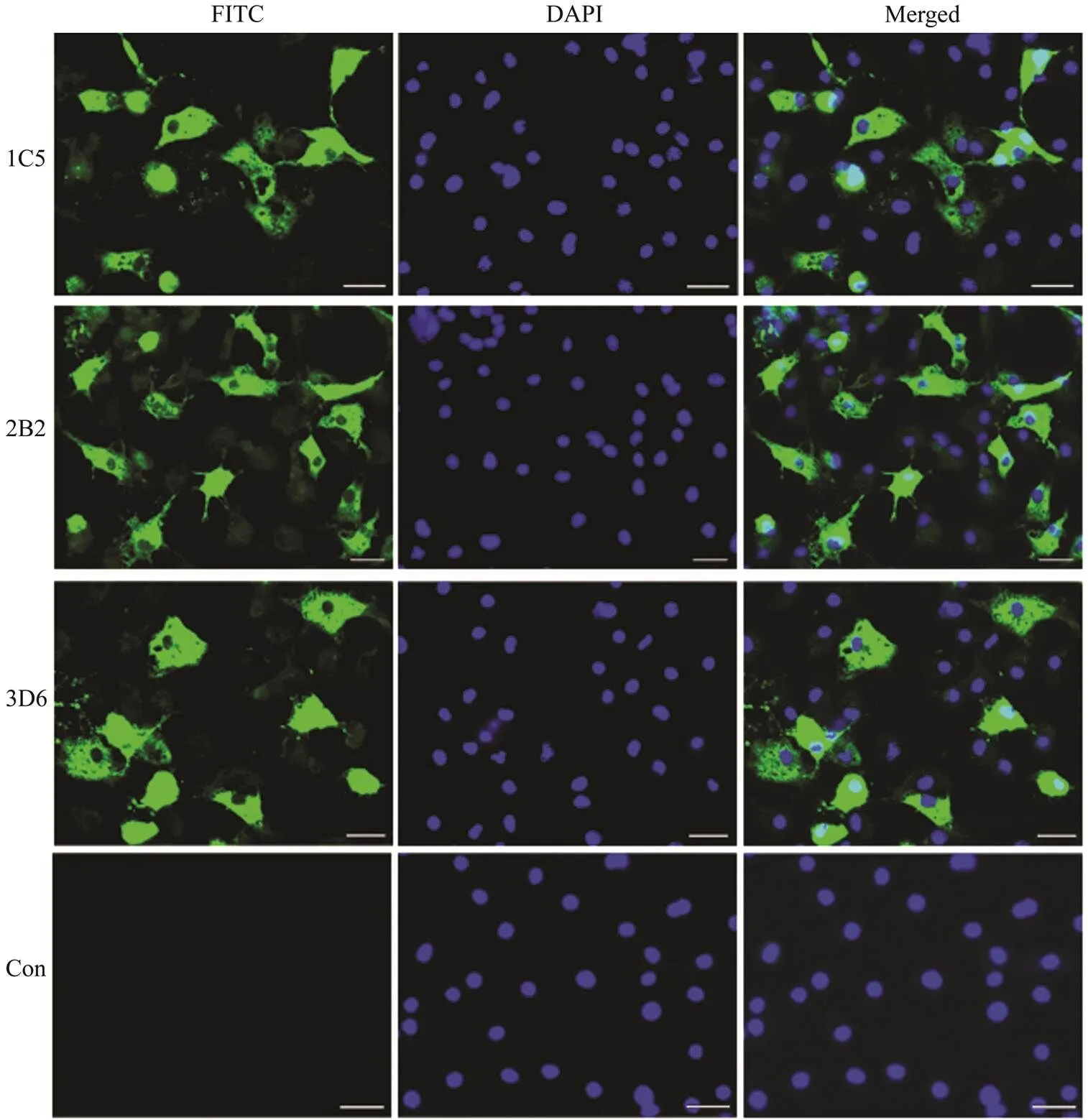
Fig.4 Immunofluorescence assay of RGNNV-infected GS cells with 3 MAbs (1C5, 2B2 and 3D6). Nuclei of cells were stained with DAPI, showing blue fluorescence. Green fluorescence signals in the cytoplasm or nuclei of infected cells in- dicate the existence of RGNNV. Scale bars=10μm.
In conclusion, three MAbs against RGNNV coat pro- tein were developed and characterized in this work. Indi- rect ELISA, western blotting and IFA results showed that these MAbs can specifically recognize the nature Cp in RGNNV and be used to develop a rapid detection method. This study provides a foundation for further studies on the rapid diagnosis of RGNNV and its infection mechanisms.
Acknowledgements
This study was supported by the National Natural Sci- ence Foundation of China (No. 31972834), and the National Key Research and Development Program of China (No. 2018YFD0900505).
Bandín, I., and Souto, S., 2020. Betanodavirus and VER disease: A 30-year research review., 9 (2): 106.
Bradford, M. M., 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding., 72 (1-2): 248-254.
Chen, N. C., Yoshimura, M., Guan, H. H., Wang, T. Y., Misumi, Y., Lin, C. C.,., 2015. Crystal structures of a piscine be- tanodavirus: Mechanisms of capsid assembly and viral infec- tion., 11 (10): e1005203.
Chen, W. J., Yi, L. Z., Feng, S. S., Zhao, L. J., Li, J., Zhou, M.,., 2017. Characterization of microRNAs in orange-spottedgrouper () fin cells upon red-spotted grou- per nervous necrosis virus infection., 63: 228-236.
Chien, M. H., Wu, S. Y., and Lin, C. H., 2018. Oral immunizationwith cell-free self-assembly virus-like particles against orange- spotted grouper nervous necrosis virus in grouper larvae,., 197: 69-75, DOI: 10.1016/j.vetimm.2018.01.012.
Doan, Q. K., Vandeputte, M., Chatain, B., Morin, T., and Allal, F., 2017. Viral encephalopathy and retinopathy in aquaculture: A review., 40 (5): 717-742.
Guo, Y. X., Chan, S. W., and Kwang, J., 2004. Membrane associa- tion of greasy grouper nervous necrosis virus protein A andcharacterization of its mitochondrial localization targeting sig- nal., 78 (12): 6498-6508.
Guo, Y. X., Dallmann, K., and Kwang, J., 2003. Identification of nucleolus localization signal of betanodavirus GGNNV protein α., 306 (2): 225-235.
Hassantabar, F., Zorriehzahra, M. J., Firouzbakhsh, F., and Thomp- son, K. D., 2021. Development and evaluation of colloidal goldimmunochromatography test strip for rapid diagnosis of ner- vous necrosis virus in golden grey mullet ()., 44 (6): 783-791, DOI: 10.1111/jfd. 13302.
Huang, Y. H., Huang, X. H., Cai, J., OuYang, Z. L., Wei, S. N., Wei,J. G.,., 2015. Identification of orange-spotted grouper () interferon regulatory factor 3 involved in antiviral immune response against fish RNA virus., 42 (2): 345-52.
Lin, C. F., Jiang, H. K., Chen, N. C., Wang, T. Y., and Chen, T. Y., 2018. Novel subunit vaccine with linear array epitope protect giant grouper against nervous necrosis virus infection., 74: 551-558.
Mori, K. I., Nakai, T., Muroga, K., Arimoto, M., Mushiake, K., and Furusawa, I., 1992. Properties of a new virus belonging to nodaviridae found in larval striped jack () with nervous necrosis., 187 (1): 368-371.
Nishizawa, T., Gye, H. J., Takami, I., and Oh, M. J., 2012. Poten- tiality of a live vaccine with nervous necrosis virus (NNV) for sevenband grouperat a low rear- ing temperature., 30 (6): 1056-1063, DOI: 10.1016/j. vaccine.2011.12.033.
Pakingking, R. J., Bautista, N. B., Jesus-Ayson, E. G., and Reyes, O., 2010. Protective immunity against viral nervous necrosis (VNN) in brown-marbled grouper () following vaccination with inactivated betanodavirus., 28 (4): 525-533, DOI: 10.1016/j.fsi.2009. 12.004.
Qin, Y. H., Liu, J. Y., Liu, W. M., Shi, H. R., and Liu, X. Q., 2020.First isolation and identification of red-grouper nervous necro- sis virus (RGNNV) from adult hybrid hulong grouper (×) in China., 529: 735662.
Tang, X. Q., Qin, Y. H., Sheng, X. Z., Xing, J., and Zhan, W. B., 2018. Generation, characterization and application of mono- clonal antibodies against matrix protein of hirame novirhabdo-virus (HIRRV) in flounder., 128: 203-213.
Valero, Y., Awad, E., Buonocore, F., Arizcun, M., Esteban, M. Á.,Meseguer, J.,., 2016. An oral chitosan DNA vaccine againstnodavirus improves transcription of cell-mediated cytotoxicity and interferon genes in the European sea bass juveniles gut and survival upon infection., 65: 64-72, DOI: 10.1016/j.dci.2016.06.021.
Wang, X. J., and Zhan, W. B., 2006. Development of an immu- nochromatographic test to detect White Spot Syndrome Virus of shrimp., 255 (1-4): 196-200.
Xing, J., Zhang, Z. Q., Sheng, X. Z., Tang, X. Q., Chi, H., and Zhan,W. B., 2020. Identification and characterization of a new strain of nervous necrosis virus isolated from pearl gentian grouper (×) in Chi- na., 529: 735663.
Zhong, Y., Tang, X. Q., Sheng, X. Z., Xing, J., and Zhan, W. B., 2019. Voltage-dependent anion channel protein 2 (VDAC2) and receptor of activated protein C kinase 1 (RACK1) act as functional receptors for lymphocystis disease virus infection., 93: e00122-19.
December 25, 2020;
February 19, 2021;
July 6, 2021
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2022
. Tel: 0086-27-87282113
E-mail: xueqinliu@mail.hzau.edu.cn
(Edited by Qiu Yantao)
杂志排行

Journal of Ocean University of China的其它文章
- The Role of Bottom Currents on the Morphological Development Around a Drowned Carbonate Platform, NW South China Sea
- Controls on the Gas Hydrate Occurrence in Lower Slope to Basin-Floor, Northeastern Bay of Bengal
- Effective Elastic Thickness of the Lithosphere in the Mariana Subduction Zone and Surrounding Regions and Its Implications for Their Tectonics
- Geophysical Evidence for Carbonate Platform Periphery Gravity Flows in the Xisha Islands, South China Sea
- Seismic Characteristics and Hydrocarbon Accumulation Associated with Mud Diapir Structures in a Superimposed Basin in the Southern South China Sea Margin
- Simulation and Analysis of Back Siltation in a Navigation Channel Using MIKE 21