豆类活性肽PA1b在大肠杆菌中的多拷贝串联表达
2022-08-11黄欣媛邹礼平范红波
黄欣媛,邹礼平,范红波
(1.湖北工程学院湖北省植物功能成分利用工程技术研究中心,湖北孝感 432000;2.湖北职业技术学院医学基础部,湖北孝感 432000)
PA1b(pea albumin 1 subunit b,豌豆白蛋白1b,又名豌豆胰岛素、aglycin)是在豌豆种子中分离鉴定出的一种由37个氨基酸残基组成的小分子活性肽,在其他豆科植物中有多种同源异构体广泛存在。它们分子内均含有3对二硫键,形成高度紧凑的空间结构,因而性质非常稳定,耐杀菌剂变性,也不被胃蛋白酶和胰蛋白酶降解。PA1b具有抗虫活性,能杀死象鼻虫、蚜虫、蚊虫等害虫,而对蜜蜂等益虫无害,有望开发成新型生物杀虫剂。PA1b还能改善糖尿病模型鼠的糖脂代谢,具有开发成口服降糖多肽药物的潜力。
基于PA1b在农业病虫害防治和糖尿病药物开发中的潜力,大量获取PA1b蛋白,进行工业规模应用或深入研究其作用机制就显得十分必要。虽然可通过从豆科植物种子中提取纯化来制备PA1b,但需避免同源异构体混入、操作繁琐、成本高且产量低下,最理想的还是通过基因工程技术将PA1b在异源系统中进行表达。目前,在大肠杆菌表达系统中对小片段的基因进行串联表达,以提高其蛋白表达量的方法已成功应用于多种小分子肽的原核表达制备,被证实简便且有效。本研究利用同尾酶法构建了多拷贝串联基因,置于表达载体pET32a(+)上,在大肠杆菌BL21(DE3)中进行了诱导表达,并用肠激酶对融合标签和串联体进行切割,获得重组PA1b多肽,为PA1b工业化应用提供参考。
1 材料与方法
1.1 材料
质粒pET32a(+)、大肠杆菌BL21(DE3)和DH5α,由笔者所在实验室保存。
1.2 主要试剂和仪器
限制性内切酶(Ⅰ、H Ⅰ和Ⅱ)、TDNA连接酶,购自Thermo Fisher Scientific;小量质粒提取试剂盒、DNA胶回收试剂盒、SanPCR扩增试剂盒、异丙基硫代---半乳糖苷(IPTG)、Bradford蛋白定量试剂、蛋白质Marker、Ni-NTA琼脂糖亲和纯化树脂预装柱、肠激酶(带His标签),均购自生工生物工程(上海)股份有限公司;超滤管、透析袋为Millipore产品;其他常用试剂均为国产分析纯。
PCR仪,东胜创新生物科技有限公司;超声波细胞粉碎机,宁波新芝生物科技股份有限公司;超微量分光光度计,美析(中国)仪器有限公司;核酸蛋白检测仪、核酸电泳仪、垂直电泳仪,北京六一生物科技有限公司。
1.3 PA1b基因合成和载体设计
由图1可知,将PA1b的氨基酸序列(A S C N G V C S P F E M P P C G S S A C R C I P V G L V V G Y C R H P S G)按照大肠杆菌的密码子偏好性进行优化,转变成DNA序列,同时在5′端添加“HⅠ酶切位点(GGATCC)+肠激酶酶切位点(GACGACGACGACAAG)”,3′端添加“Ⅱ酶切位点(AGATCT)+肠激酶酶切位点(GACGACGACGACAAG)+Ⅰ酶切位点(C T C G A G)”,交由生工生物工程(上海)股份有限公司合成该基因片段并置于pUC57载体上,命名为pUC57-1PA1b。该序列中,HⅠ和Ⅰ酶切位点是为了使用双酶切将基因片段亚克隆到表达载体pET32a(+)上;HⅠ和Ⅱ酶切位点是为了使用同尾酶法构建串联重组载体;肠激酶酶切位点是为了在融合蛋白分离纯化以后使用肠激酶进行特异性切割,将硫氧还蛋白(Trx A)、6×His等标签去除,同时也将串联的PA1b剪切成单个多肽单元。

1.4 重组表达载体pET32a(+)-nPA1b的构建
首先,采用同尾酶法构建PA1b串联多拷贝重组质粒pUC57-(=2,3,4),方法见图2,重复该过程,可依次获得基因串联重复2、3、4次的重组质粒。分别采用CaCl法转化入感受态大肠杆菌DH5α中,菌落PCR鉴定阳性克隆的正确性。

其次,将pUC57-载体上的基因片段亚克隆至表达载体pET32a(+)上。方法是将验证后得到的pUC57-质粒(=1,2,3,4)采用HⅠ与Ⅰ双酶切切下串联基因片段,胶回收该片段,与同样双酶切纯化后的pET32a(+)载体片段按照摩尔浓度比8∶1混合,TDNA连接酶16 ℃过夜连接。连接产物用CaCl法转化入BL21(DE3)感受态大肠杆菌,从含氨苄青霉素的LB平板上挑取菌落进行培养,小量抽提质粒,以pET32a(+)质粒上多克隆位点上游的通用引物SoTag primer(G A A C G C C A G C A C A T G G A C A G C)和下游的通用引物T7 terminator primer(G C T A G T T A T T G C T C A G C G G)进行PCR验证,将阳性转化子送至上海生工生物工程有限公司测序。鉴定正确的质粒命名为pET32a(+)-(=1~4)。
1.5 重组PA1b的诱导表达
挑取含pET32a(+)-的大肠杆菌单菌落,接种于含氨苄青霉素(100 μg/mL)LB液体培养基中,37 ℃培养过夜,再按100倍稀释比例转接至新鲜含100 μg/mL氨苄青霉素的LB液体培养基中,37 ℃培养至为0.6时,加入1 mmol/L的IPTG,25 ℃下诱导培养8 h。同时,设置1组pET32a(+)空质粒转化菌作为对照组。
1.6 表达产物的SDS-PAGE检测
诱导结束后,4 ℃离心收集菌体,PBS洗涤2次,PBS重悬后冰浴中进行超声破壁(功率400 W,总时间20 min,工作2 s,间隔6 s),4 ℃下12 000 r/min离心15 min,收集上清,将沉淀用包涵体溶解液(8 mol/L 尿素,50 mmol/L Tris-HCl,300 mmol/L NaCl,0.1% Triton X-100,pH值8.0)溶解。Bradford法测定上清和包涵体溶解液的总蛋白浓度,取25 μg总蛋白上样,进行12%分离胶的SDS-PAGE,考马斯亮蓝染色,凝胶成像仪记录电泳图谱。分析PA1b融合蛋白的可溶性,采用ImageJ v1.52图像软件分析条带的光密度,计算PA1b蛋白的相对含量。
1.7 PA1b融合蛋白的亲和纯化
按“1.5”节和“1.6”节方法进行诱导表达和菌体破碎,离心收集包涵体,包涵体用柱平衡缓冲液(20 mmol/L Tris-HCl,500 mmol/L NaCl,5 mmol/L咪唑,8 mol/L尿素,pH值8.0)重溶后通过 Ni-NTA 琼脂糖树脂柱进行亲和层析纯化。进样前,利用2倍柱体积的平衡缓冲液平衡柱子。进样后,采用5~10倍柱体积的平衡缓冲液清洗柱子,直至流穿液的接近基线。最后采用2~5倍柱体积的洗脱缓冲液(20 mmol/L Tris-HCl,500 mmol/L NaCl,250 mmol/L咪唑,8 mol/L尿素,pH值8.0)洗脱带有6×His标签的PA1b融合蛋白。透析去除咪唑和尿素,超滤管离心浓缩获得PA1b融合蛋白。
1.8 融合蛋白的肠激酶酶切和纯化
将纯化的PA1b融合蛋白置于肠激酶酶切缓冲液(25 mmol/L Tris-HCl,50 mmol/L NaCl,2 mmol/L CaCl,pH值7.6),按照50 μg融合蛋白加1U肠激酶的用量在16 ℃酶切36 h。酶切后的混合液再次进行Ni-NTA琼脂糖树脂亲和层析纯化,切下的Trx-His标签和带有His标签的肠激酶结合到镍柱上,而PA1b多肽存在于流穿液中。收集流穿液,采用截留量10 ku超滤管纯化PA1b多肽,PA1b多肽在超滤管离心后的透过液中,进行Tricine SDS-PAGE检测。
2 结果与分析
2.1 表达载体pET32a(+)-nPA1b的构建
PA1b由111 bp DNA序列编码,在基因合成时两端分别添加了HⅠ和Ⅱ的酶切序列,由图2可知,采用同尾酶法构建了串联重复序列载体pUC57-(=2,3,4),再亚克隆至表达载体pET32a(+)上,构建成pET32a(+)-(图1),转化大肠杆菌后挑取阳性克隆,采用SoTag和T7 terminator引物进行PCR扩增插入片段,由图3可知,显示扩增出的条带与目标片段预期大小(PA1b单拷贝320 bp,二拷贝452 bp,三拷贝 584 bp,四拷贝716 bp)一致。进而进行DNA测序验证,结果发现插入片段的插入位置正确,且序列与理论设计完全一致,说明多拷贝PA1b基因串联载体构建成功。

2.2 PA1b融合蛋白的诱导表达和可溶性分析
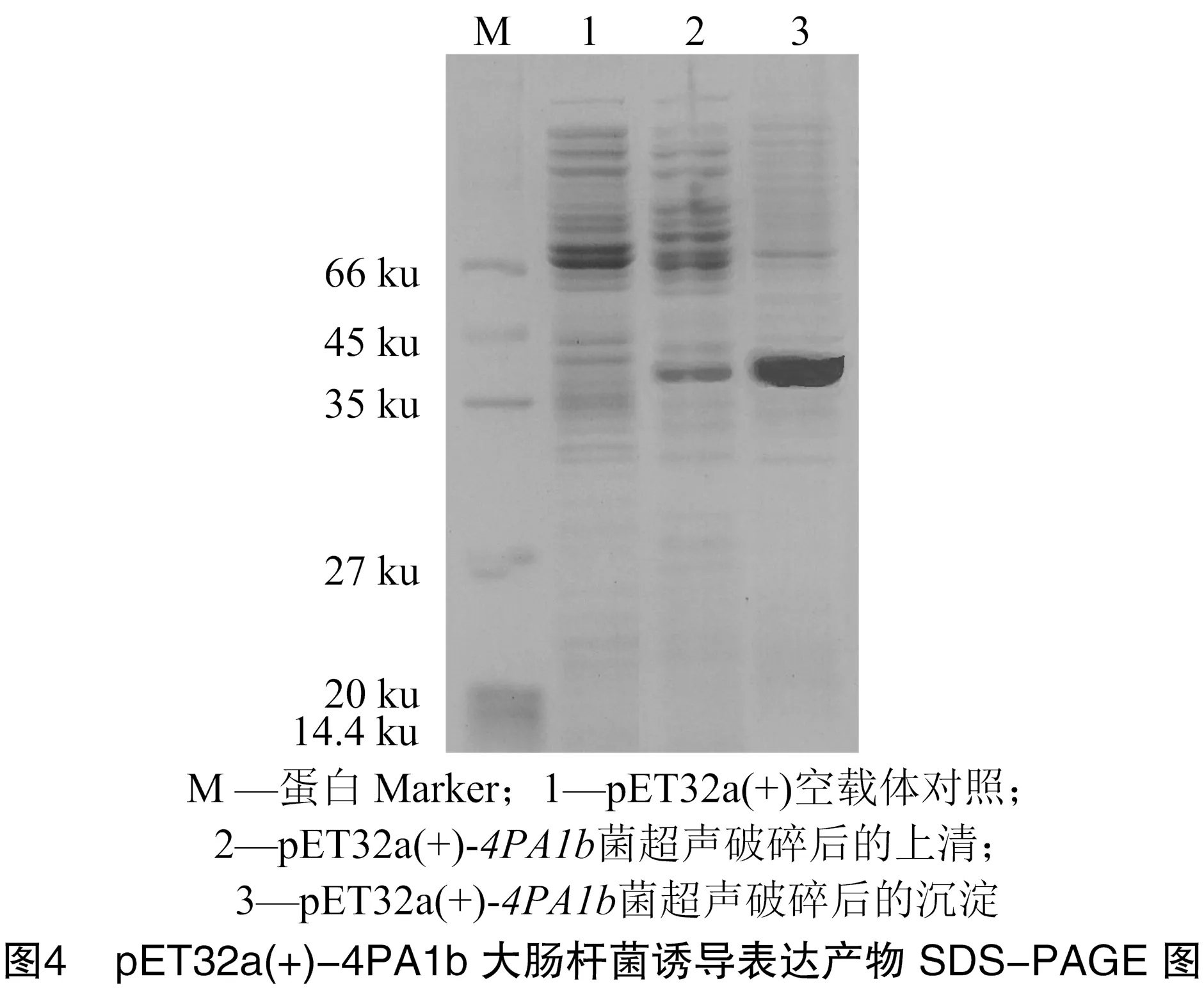
IPTG诱导pET32a(+)-宿主菌表达后,超声破碎菌体,SDS-PAGE分析破碎组分的上清和沉淀部分,由图4可知,相对于空载体对照,pET32a(+)-菌体裂解液上清和沉淀部分均明显多出1个约38 ku条带,和4拷贝串联PA1b融合蛋白理论分子量相符,说明融合蛋白诱导表达成功,且主要表达在包涵体中。
2.3 不同拷贝数PA1b融合蛋白的SDS-PAGE检测
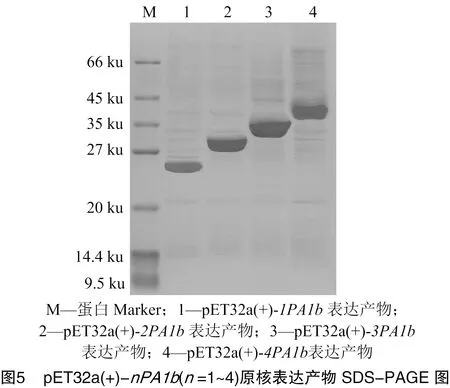
采用IPTG分别对含1~4串联重复基因pET32a(+)-质粒的大肠杆菌进行诱导,SDS-PAGE电泳检测表达产物,由图5可知,1~4拷贝数PA1b融合蛋白的理论分子量分别约为25.2、29.4、33.6、37.8 ku,研究观察到各融合蛋白分子量与理论分子量相符,说明pET32a(+)-(=1,2,3,4)原核表达载体均能成功诱导表达。采用Image J软件对各泳道进行光密度分析,由表1可知,发现各拷贝数PA1b融合蛋白在包涵体总蛋白中的相对含量有差异,换算成PA1b的相对含量也有差异。理论上,质粒中基因拷贝数越多,与载体质量比越大,PA1b在融合蛋白中的含量就越高。根据图5中融合蛋白实际表达水平进行换算,结果发现,pET32a(+)-表达产物中PA1b的相对含量最高(23.3%),约为单拷贝(5.5%)的4.2倍,因此在后续的融合蛋白分离纯化和酶切试验中均选用pET32a(+)-菌株作为诱导表达菌。3拷贝和4拷贝串联表达产物中PA1b的相对含量几乎相同,说明蛋白表达量和拷贝数之间并非线性正相关。
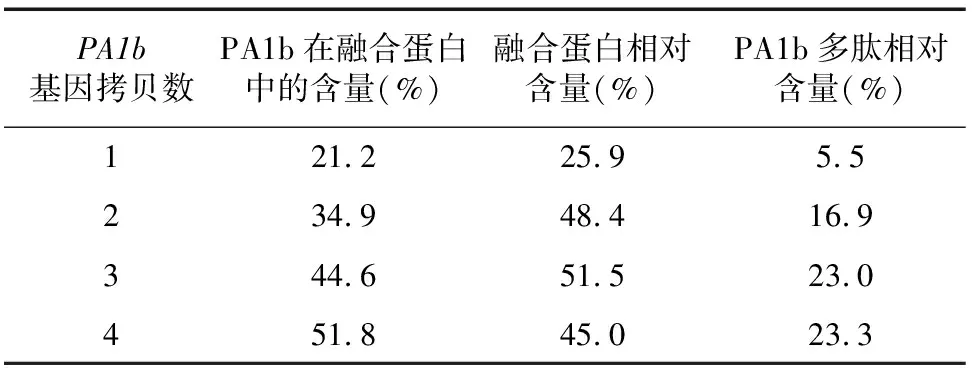
表1 pET32a(+)-nPA1b表达融合蛋白水平及其中PA1b相对含量
2.4 PA1b融合蛋白的纯化
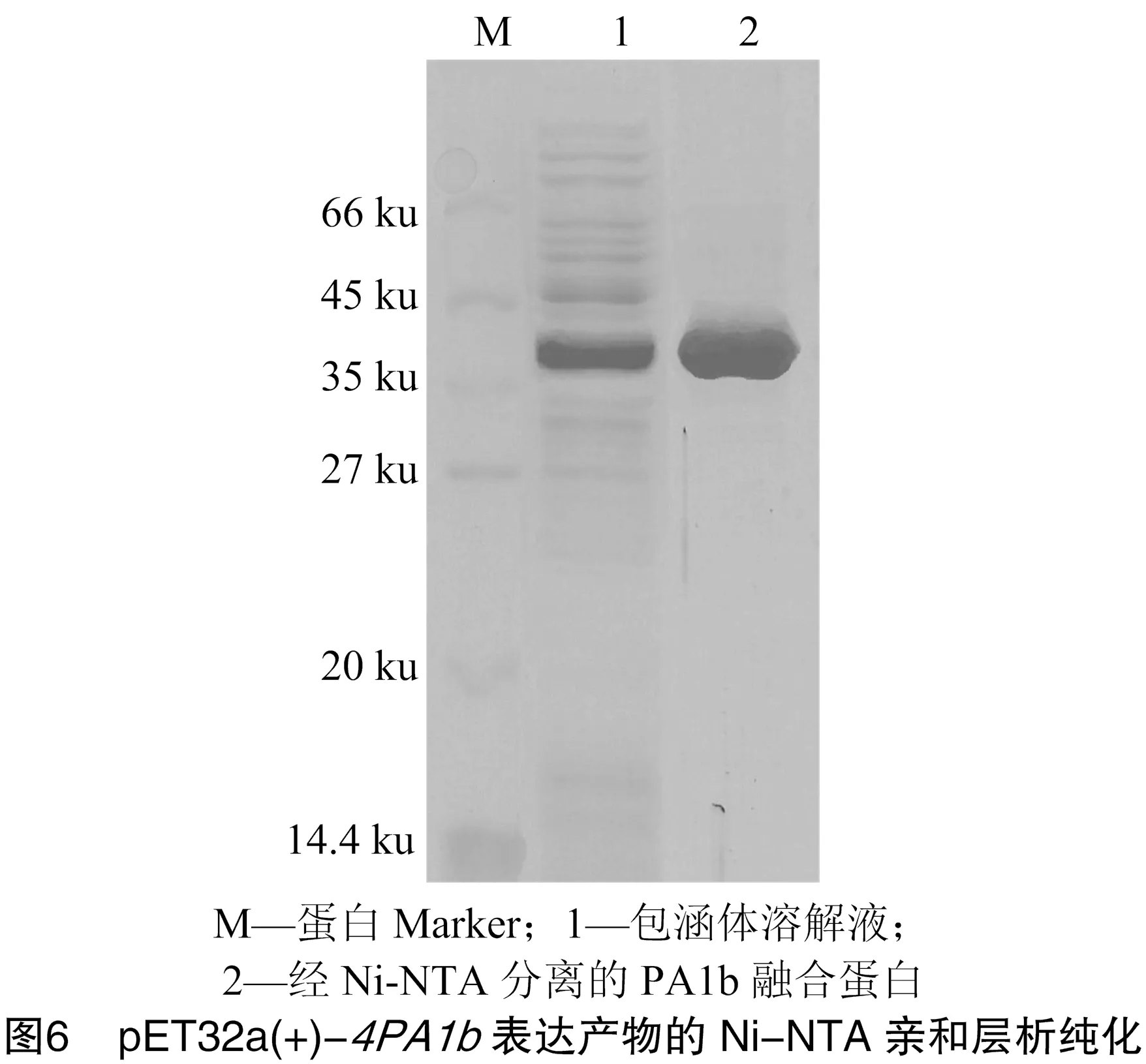
将pET32a(+)-的诱导表达产物进行Ni-NTA柱亲和层析纯化,SDS-PAGE检测结果,由图6可知,发现分离纯化后的4拷贝PA1b融合蛋白具有较高纯度(97.8%)。
2.5 PA1b融合蛋白肠激酶酶切
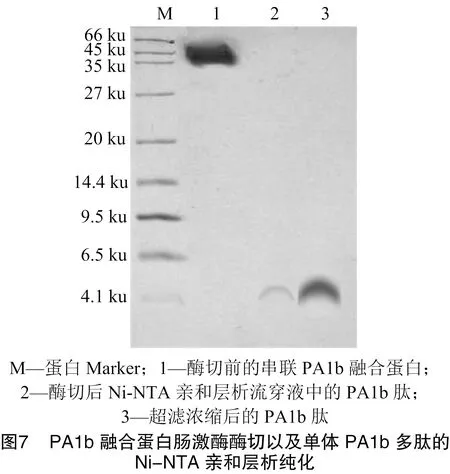
由图7可知,将纯化后的PA1b融合蛋白采用肠激酶酶切,完全酶切后的蛋白混合液经过Ni亲和层析纯化后,流穿液中得到切开的无TrxA标签、无6×His标签的单拷贝PA1b多肽,经超滤管浓缩去杂后,Tricin-SDS-PAGE检测分子量约为4.5 ku,与理论预期相符。
3 讨论与结论
大肠杆菌表达系统具有高效、条件易控、成本低等优点,是当前应用最广泛的蛋白表达系统之一。因此,本研究选用大肠杆菌作为PA1b的表达宿主。PA1b是一种小分子量多肽,且富含半胱氨酸,因此在进行原核表达时,要考虑如何提高蛋白表达量、促进分子内二硫键的正确形成、保持多肽天然折叠构象等因素。本研究采用构建串联多拷贝基因的策略来提升表达量,通过同尾酶法构建了插入1~4串联拷贝数PA1b的重组载体,分别转化大肠杆菌后诱导表达,SDS-PAGE分析了各融合蛋白的表达情况,结果发现在“1.5”节中所述的诱导条件下,PA1b融合蛋白主要存在于包涵体中,且多拷贝数PA1b的表达量高于单拷贝,4拷贝串联PA1b蛋白含量最高,提示串联表达是提高产量的有效方法。然而,拷贝数并非越多越好,最佳拷贝数仍取决于载体上串联肽与融合标签的相互影响、实验诱导条件等多种因素。
本研究将串联基因插入到具有Trx A、6×His等多个融合标签的pET32a(+)载体上进行融合表达,Trx A能提供氧化还原动力,促进PA1b分子内二硫键形成。考虑到pET32a(+)载体上标签的去除可采用肠激酶酶切法,所以在构建串联基因时,在各拷贝间也添加了肠激酶酶切位点。因此,在融合蛋白亲和纯化后,采用肠激酶酶切,既可去掉TrxA和6×His融合标签,也能将串联的PA1b切割成单体重组PA1b多肽。结果表明,pET32a(+)-的诱导表达产物经过亲和纯化后可获得高纯度的4拷贝PA1b融合蛋白,再经肠激酶酶切、亲和层析去杂、超滤浓缩后获得了不带TrxA标签的重组PA1b多肽。它比天然PA1b的C末端多了7个氨基酸残基:Arg-Ser-Asp-Asp-Asp-Asp-Lys,这是否会对PA1b的活性造成影响,有待后续研究。
在PA1b的原核表达方面,国内外也有报道。2003年,Hanada等在BL21大肠杆菌中导入leginsulin(PA1b在黄豆中的同源异构体,同源性约64%)基因,表达出带有Trx标签的leginsulin。2004年,杜雯等将PA1b与麦芽糖结合蛋白(MBP)构建成融合基因在大肠杆菌DH5α中进行了周质空间可溶性表达,然而融合蛋白的结构过于庞杂而无PA1b活性,表达量太低,及MBP标签去除需经Factor Xa酶切成本高,因而并无后续。最近,黄敏华等利用内含肽与自组装肽与Aglycin(PA1b别名)连接,在大肠杆菌BL21(DE3)中进行诱导表达,得到目标蛋白聚集体,再通过内含肽介导的N端自剪切,去掉了融合标签,制备无标签的Aglycin多肽。
综上,本研究采用串联表达的方案在大肠杆菌表达系统中制备了豆类活性肽PA1b的4拷贝融合蛋白,有效提高了PA1b表达量。经肠激酶切割后,得到了无标签的单拷贝PA1b重组多肽,为多拷贝法制备PA1b肽奠定了基础。
