氯化物熔盐中铬的价态对镍基合金腐蚀性的影响
2022-08-10魏小兰戚文杰丁静陆建峰王维龙刘书乐
魏小兰,戚文杰,丁静,陆建峰,王维龙,刘书乐
(1 华南理工大学化学与化工学院,广东 广州 510640; 2 中山大学材料科学与工程学院,广东 广州 510006)
引 言
近年来全球能源消费总量迅速增长,同时全球气候变化加剧各国对清洁能源的需求。聚光太阳能热发电技术(concentrated solar power,CSP)是一种有前景的清洁能源利用技术[1]。通过与大规模的储热系统相结合,CSP 发电站可一定程度上克服太阳能利用过程中所面临的间歇性问题,从而提供平稳可调峰的电力。目前CSP发电站的储热系统多使用硝酸盐熔盐作为传热储热的工质,但由于硝酸熔盐在高温下受热易分解,并产生对环境有害的氮氧化物,因此限制了系统的最高工作温度[2]。为满足商业化需求,CSP发电站须提高运行的工作温度,以实现更低的单位发电成本以及更高的能源利用效率。而氯化物熔盐在高温下有更好的稳定性,且具有储热性能好、原料存量大、成本低廉等优势,有望成为下一代CSP储热系统的传热储热流体[3-4]。
高温氯化物熔盐对金属结构材料的腐蚀是制约其应用的重要因素[5-6]。在高温氯化物熔盐储热系统中,富铬(Cr)合金结构材料会受到侵蚀性腐蚀,主要表现为合金表面与晶界中Cr 的优先流失[7-11],进而使熔盐熔体中Cr的含量上升。研究表明,腐蚀后的氯化物熔盐中,Cr 含量可达几十到数千ppm(1 ppm=1 mg/L)[12-13],其具体的浓度取决于腐蚀时间、温度、合金中的Cr含量等。
从合金进入熔盐中的Cr 元素可以是多种价态的。美国橡树岭国家实验室研究发现[14],在氟盐中,由金属进入盐中铬价态有Cr2+和Cr3+两种,二者含量与氟熔盐酸碱性相关。Bawane 等[15]通过电子能量损失谱的研究发现,氩气气氛下,铬在ZnCl2以及KCl-MgCl2熔盐中腐蚀后的价态为+3。王学良[16]研究金属铬在NaCl-KCl熔盐中腐蚀时发现,铬还会以0价态的形式微量扩散于熔盐中。
在实际工况下,熔盐在合金管道中冷热循环流动,由于温度的改变,Cr 的溶解度亦会发生变化,从而可能发生局部的积累。这种溶解于熔盐中的Cr元素一定程度改变了原有熔盐的组分,对熔盐的热物性或腐蚀性可能造成影响。如彭浩[17]在LiFNaF-KF 熔盐中分别引入CrF2和CrF3后发现,Cr3+因其具有一定氧化性会加剧对316L不锈钢的腐蚀,而Cr2+则有效抑制腐蚀。阴慧琴[18]为研究进入熔盐的腐蚀产物对LiF-NaF-KF 熔盐热物性的影响而引入6000 ppm CrF3,结果发现CrF3对热物性影响较小,但会加速对Hastelloy-N 的腐蚀。但这些研究主要集中在氟化物熔盐中。在氯化物熔盐中Cr 的溶解亦引起了一些研究者的关注,如通过第一性原理分子动力学研究Cr 在氯化物熔盐中的微观结构和扩散情况[19-20],但对熔盐腐蚀性的影响研究较少。
已有的实验研究表明,在氯化物熔盐中,铁基合金耐腐蚀性不佳,而镍基合金则相对较好[7],为此镍基合金可以作为储热系统中换热器等关键部位的结构合金。而其中哈氏合金(Hastelloy alloy)是应用较为广泛的商用镍基合金,一般而言具有良好的抗腐蚀性和热稳定性[21]。为了解进入氯化物熔盐中不同价态铬对金属后续腐蚀性的影响,本研究在三元NaCl-MgCl2-CaCl2共晶熔盐中分别引入可能的腐蚀产物Cr、CrCl2、CrCl3,探讨不同含铬熔盐对一种贫铬Hastelloy B-2(HB-2)和两种富铬Hastelloy C-276(HC-276)、Hastelloy X(HX) 哈氏镍基合金在高温下的腐蚀行为。通过测量腐蚀前后合金试样的质量损失、分析合金试样表层和剖面的微观形貌与组成,从而研究进入熔盐中铬的价态对三种合金腐蚀性的影响。
为了揭示进入熔盐中不同价态的铬对金属腐蚀性影响的机理,计算了CrCl3分别氧化单质Cr、Fe、Ni、Mo反应的标准Gibbs自由能,根据其代数值判断CrCl3在熔盐腐蚀中的作用。
1 实验材料和方法
1.1 实验材料
研究所用镍基合金HB-2、HC-276 与HX 的化学组成如表1 所示。将三种合金切割成30 mm×15 mm×2 mm 尺寸的试样。腐蚀实验之前,依次用74.0~10.0 μm 颗粒度的SiC 砂纸打磨试样表面,以除去油污及氧化层;再分别用粒径为2.5 μm 和1.0 μm 金刚石抛光剂将试样抛光至镜面;经去离子水、无水乙醇清洗后置于90℃烘箱中烘干备用。使用分析天平(T-114,北京赛多利斯天平有限公司)称量每片试样的质量。

表1 三种镍基合金的化学组成Table 1 Chemical composition of three nickel base alloys
以市售NaCl(纯度≥99.5%,广州化学试剂厂)、MgCl2(纯度≥99%,上海麦克林生化科技有限公司)、CaCl2(纯度≥96%,天津市大茂化学试剂厂)按最低共熔点组成NaCl-MgCl2-CaCl2(39.5%∶39.1%∶21.4%)[22]制备三元共晶熔盐。熔盐的制备及腐蚀实验采用的容器均为刚玉坩埚,其在氯化物熔盐相关研究中亦较多采用[8,23-24]。首先,将三种原料盐按比例机械混合于坩埚中,将其置于马弗炉中于250℃下恒温3 h以除去盐中的吸附水,再升温至600℃并恒温3 h使之形成均匀混合流体。然后冷却至室温,经研磨粉碎即得到三元NaCl-MgCl2-CaCl2熔盐作为基础盐(base salt),记为BS。
1.2 实验及表征方法
将所制的NaCl-MgCl2-CaCl2基础盐分别置于四个编号为1~4的刚玉坩埚中于高温下熔化。分别向编号为2、3、4 号的坩埚加入0.4%Cr(纯度≥99.5%),0.95%CrCl2(纯度≥97%)和1.22%CrCl3(纯度≥98%),使加入的铬元素的摩尔比例一致,以制备含Cr0、Cr2+以及Cr3+的熔盐,分别命名为BS-Cr0、BS-Cr2+和BSCr3+。所加入的Cr、CrCl2和CrCl3均购自上海麦克林生化科技有限公司。
将BS 分别装入3 个200 ml 的刚玉坩埚中,然后分别浸入抛光的试样HB-2、HC-276 和HX,盖上盖子以减少外界气氛对体系的影响。分别用BS-Cr0、BS-Cr2+和BS-Cr3+盐代替BS 进行相同操作。然后将上述12 个坩埚同时置于马弗炉中,600℃下恒温腐蚀时间为100 h。腐蚀实验结束后,从坩埚中取出试样,经腐蚀后试样分别用去离子水超声清洗,以去除残留的熔盐。称量腐蚀后合金的质量,计算单位面积的质量损失,如式(1)所示。

式 中,m1为 试 样 的 原 始 质 量,mg;m2为 腐 蚀100 h 后的质量,mg;S0为试样的原始表面积,mm2。以质量损失率表征试样表观腐蚀程度。
采用X 射线衍射(XRD,德国Bruker D8 Advance)对腐蚀前后的试样进行物相分析,通过扫描电镜(SEM,日本日立3700N 型)分析腐蚀前后试样表面和截面的形貌,同时利用能量色散光谱仪(EDS)表征试样表面元素分布。
2 实验结果与讨论
2.1 质量损失
三种合金HB-2、HC-276 和HX 分别在BS 盐,以及BS-Cr0、BS-Cr2+和BS-Cr3+盐中600℃下腐蚀100 h 后的质量损失率,如图1 所示。试样质量的变化受两方面的影响:一是合金元素发生一定程度的流失,会导致质量降低;二是合金表面腐蚀氧化层的形成,会导致质量的上升。由图1可见,所有金属的质量对比腐蚀前都有所降低,但降低的幅度不一,意味着在该温度下三种合金在氯化物熔盐中出现了程度不一的腐蚀,且腐蚀行为主要表现为合金中元素的流失。
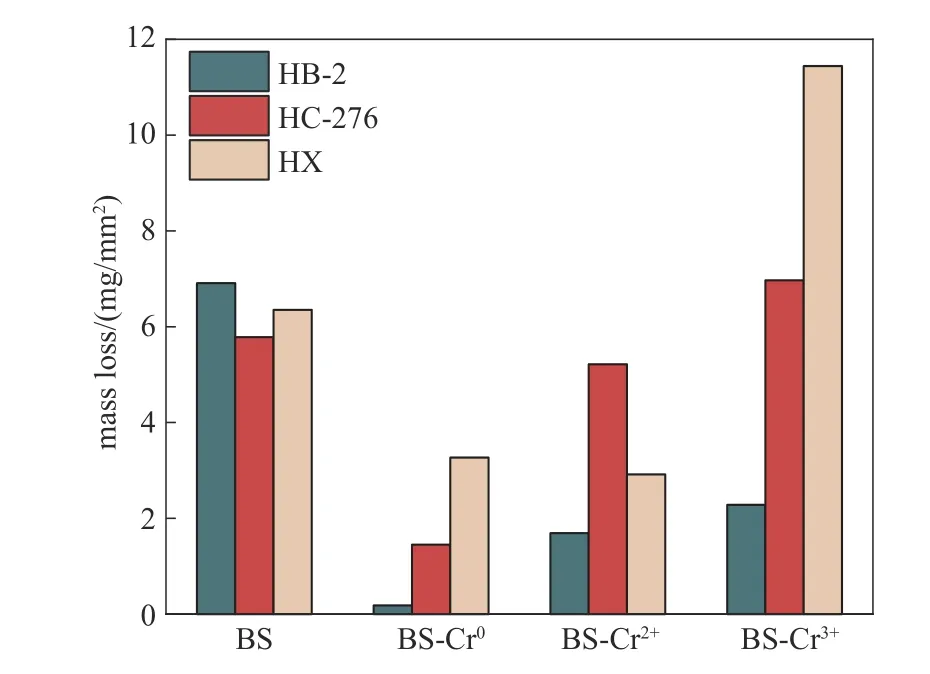
图1 HB-2、HC-276和HX在四种熔盐中腐蚀100 h后的质量损失Fig.1 Mass loss of HB-2,HC-276 and HX after corrosion in four molten salts for 100 h
与在BS 中的腐蚀情况对比,在BS-Cr0以及BSCr2+熔盐中,三种合金的质量损失均有所降低。而在BS-Cr3+中,HC-276与HX 的质量损失上升,HB-2质量损失下降。且对于三种合金,其质量损失均随着所引入铬的价态增大而上升。
根据表面竞争吸附理论[25],在氯化物熔盐中的大量氯离子竞争作用下,氧化物层生成往往较为缓慢。且高温下增强了原子动能涨落,使得合金中的元素容易流失。以合金中元素流失导致的质量变化初步判断腐蚀严重程度,可见熔盐中含有低价的Cr0和Cr2+时,能有效抑制腐蚀的质量损失。而当熔盐中引入较高价的Cr3+时,对富铬的HC-276 和HX有明显的促进腐蚀作用,这与316L不锈钢在氟化物熔盐中的腐蚀现象类似[17]。而Cr3+引入熔盐后对于贫铬的HB-2 表现出一定的抑制腐蚀效果,能减少合金元素流失。
2.2 晶体结构分析
图2是HB-2、HC-276和HX在腐蚀前后的XRD分析结果。在三种熔盐中腐蚀100 h后,三种合金均很大程度上保留了其本征物相结构。HC-276及HX在三个主峰对应更高的2θ位置产生了新峰,这可能是由于金属表面某些元素消耗导致的晶面尺寸减小引起XRD峰的偏移[8,26]。相较于在BS-Cr0中的腐蚀,在添加了Cr2+以及Cr3+的熔盐中腐蚀后样品出现了明显新峰,表明腐蚀后的HC-276、HX合金表面在X射线可及的深度,可能出现了新的物相。图2中HB-2与HC-276 中均没有出现明显的表征氧化物腐蚀产物的物相,这可能与一般的金属氧化物在氯化物熔盐中不稳定有关[27];或是氧化物腐蚀层太薄,在腐蚀实验中没有形成附着稳定的氧化物层,难以收集到足够的腐蚀层信息。而HX 合金Cr、Fe 含量相对较高,氧化后容易与来源于熔盐中Mg2+水解生成的氧化镁进一步反应后产生MgCr2O4、MgFe2O4,两者在XRD上较难分辨。结合后文HX在BS-Cr3+中的线扫结果显示,在氧化层中Cr的含量相对较高,可能主要产物为MgCr2O4,该氧化物能在氯化物熔盐中生成[7,28]。

图2 HB-2、HC-276和HX在BS-Cr0、BS-Cr2+和BS-Cr3+三种熔盐中腐蚀100 h前后的XRD谱图Fig.2 XRD patterns of HB-2,HC-276 and HX before and after corrosion for 100 h in molten salts of BS-Cr0、BS-Cr2+and BS-Cr3+
2.3 腐蚀形貌
图3 是HB-2、HC-276 和HX 在BS、BS-Cr0、BSCr2+和BS-Cr3+熔盐中600℃腐蚀100 h 并超声水洗清后的表面形貌。由图3可见,在BS盐中,三种金属均遭到明显的腐蚀。对于HB-2表面,可以看出其表面整体上略凹凸不平,出现多道“沟壑”,把表面分割成多个小块,为明显的晶界腐蚀。在非晶界腐蚀的部位,在表面主要以一些细小晶粒的脱落为主,并未形成明显的腐蚀孔洞。经过100 h 的腐蚀后,HC-276表面已较不平整,存在晶界腐蚀,且分布着较多的细小的腐蚀孔洞,这是孔蚀发生的早期。而HX的平整度最差,不仅有明显的晶界腐蚀,还出现了很多腐蚀孔洞以及较大范围的金属的脱落。由于熔融状态下的氯化物熔盐为离子熔体,具有良好的导电性,其静态腐蚀反应以电化学腐蚀为主,即熔盐中的氧化性物质作为氧化剂,而合金上的元素作为还原剂被氧化为对应的离子,其中部分被氧化的合金元素离子溶解在熔盐中。蚀孔主要是由于在该处形成了活化-钝化的微电偶腐蚀电池,蚀孔内的合金元素发生阳极溶解反应,金属阳离子不断溶解,而氯化物熔盐体系中大量的穿透性较强的氯离子可穿进蚀孔维持孔内的电中性使腐蚀不断进行。

图3 HB-2、HC-276和HX在600℃的BS(a)、BS-Cr0(b)、BS-Cr2+(c)和BS-Cr3+(d)熔盐中腐蚀100 h后的表面微观形貌Fig.3 Micromorphology of surface of HB-2,HC-276 and HX after corrosion at 600℃for 100 h in BS(a),BS-Cr0(b),BS-Cr2+(c)and BS-Cr3+(d)molten salts
横向对比图3中三种合金在含不同价态Cr的熔盐中的腐蚀表面形貌,即图3(b)、(c)、(d),发现随着所添加Cr 元素的价态的上升,表面的腐蚀痕迹加深。但具体而言,不同合金的形貌仍有差异。在BS-Cr0盐中腐蚀后,HB-2 表面平整,腐蚀较轻;HC-276 表面虽比较平整,但表面的蚀孔清晰可辨;HX 表面呈现出明显的晶界蚀痕。在BS-Cr2+熔盐中腐蚀后,HB-2 呈现晶界腐蚀的迹象;HC-276 表面的蚀孔已扩大成腐蚀凹坑,但在较平的部分分布着大量蚀孔;HX表面看似平整,但其晶界腐蚀清晰可见。在BSCr3+熔盐中腐蚀后,HB-2的腐蚀程度比在BS盐中要轻,但比在BS-Cr0和BS-Cr2+熔盐中要重;HB-2、HC-276 和HX 在BS-Cr3+熔盐中的腐蚀程度随着合金中Cr含量的增大而增大,尤其在HX表面布满了蚀坑与蚀孔。总之,在熔盐中分别引入Cr0、CrCl2和CrCl3后,它们对三种金属的腐蚀严重程度表现为Cr3+>Cr2+>Cr0,这与图1所示的质量损失情况相吻合。
对三种合金表面腐蚀前、后进行主要元素的EDS分析,分析区域均取40 mm2,分析结果如表2所示。
由表2 可见,HB-2 在腐蚀前、后表面各元素含量差异不大,说明腐蚀过程中腐蚀层主要元素Ni和Mo 可能均匀消减。HC-276 和HX 被腐蚀后其Cr、Fe 元素含量下降较多,呈现Cr 和Fe 优先流失的现象,而且合金中Cr、Fe 流失程度随着熔盐中加入的铬的价态升高而增大。
图4 是HB-2、HC-276 和HX 在BS、BS-Cr0、BSCr2+和BS-Cr3+熔盐中600℃下腐蚀100 h 并超声水洗后的截面形貌。图4(a)剖面呈现裂隙,印证了图3(a)所示HB-2 和HC-276 的晶界腐蚀现象,图4(a)剖面中的蚀孔也印证了图3(a)中HX的点蚀行为。

图4 HB-2、HC-276和HX在600oC的BS(a)、BS-Cr0(b)、BS-Cr2+(c)和BS-Cr3+(d)熔盐中腐蚀100 h后的截面微观形貌Fig.4 Microstructure of cross section of HB-2,HC-276 and HX after corrosion at 600℃for 100 h in BS(a),BS-Cr0(b),BS-Cr2+(c)and BS-Cr3+(d)molten salts
比较图4(b)和图4(c)发现,HB-2 和HC-276 在BS-Cr0和BS-Cr2+盐中的腐蚀很浅,剖面平整无明显裂隙和蚀孔,但HX 剖面出现了蚀孔,图4(d)显示HX 在BS-Cr3+盐中腐蚀后腐蚀层厚度增大,而且其剖面形貌与其在BS 熔盐中腐蚀后的差别较大。图4(d)表明,HB-2 在BS-Cr3+盐中腐蚀后,其剖面出现了腐蚀的迹象,但剖面仍然比较平整,没有出现明显的蚀孔和裂隙,与图3(d)和表2 的分析一致。
2.4 元素分布
为进一步探究Cr 元素对熔盐腐蚀性的影响机理,分析了各试样的截面元素分布情况。图5~图8是HC-276 分别在BS、BS-Cr0、BS-Cr2+和BS-Cr3+熔盐腐蚀后其剖面的元素面分布图及EDS 线扫结果。
图5 线扫的结果中,HC-276 在BS 熔盐中腐蚀后,表面数微米内氧含量明显升高,且线扫(黄线)掠过的腐蚀“通道”的氧含量亦明显上升,即在氯化物熔盐中,HC-276 的腐蚀行为并不是单纯的元素流失,仍会生成一定的氧化层。从图6 可见,HC-276 在BS-Cr0中元素的损失集中于表面,且线扫结果显示,几种合金元素的含量与HC-276 腐蚀前基本一致,唯有在最初的1~2 μm 处信号下降,意味着在BS-Cr0中主要是各种元素的均匀流失,且流失的量很少,表面未有明显的氧化层生成。从图7可见,BS-Cr2+中表面元素亦基本均匀流失,在表面有一定的氧化层。而图8 中显示,HC-276 熔盐在BS-Cr3+中腐蚀后,其腐蚀层中铬、铁元素分布明显降低,特别是铬降低幅度较大,说明熔盐中引入CrCl3会促进合金中铬和铁的流失。

图5 HC-276在BS熔盐中腐蚀100 h后截面EDS线扫结果(沿黄线)及元素分布Fig.5 EDS line scanning results(along yellow line)and cross section element distribution of HC-276 after corrosion in BS molten salt for 100 h

图6 HC-276在BS-Cr0熔盐中腐蚀100 h后截面EDS线扫结果(沿黄线)及元素分布Fig.6 EDS line scanning results(along yellow line)and cross section element distribution of HC-276 after corrosion in BS-Cr0 molten salt for 100 h

图7 HC-276在BS-Cr2+熔盐中腐蚀100 h后截面EDS线扫结果(沿黄线)及元素分布Fig.7 EDS line scanning results(along yellow line)and cross section element distribution of HC-276 after corrosion in BS-Cr2+molten salt for 100 h

图8 HC-276在BS-Cr3+熔盐中腐蚀100 h后截面EDS线扫结果(黄线)及元素分布Fig.8 EDS line scanning results(along yellow line)and cross section element distribution of HC-276 after corrosion in BS-Cr3+molten salt for 100 h


通过消耗所添加的Cr0,避免了氧化性物质直接氧化合金的元素,从而减缓了腐蚀。
对于BS-Cr2+,熔盐中大量的Cr2+相当于提高了反应式(2)、式(3)中生成物的浓度,降低了Cr 被氧化的化学势,从而降低了Cr 被氧化的速率,Cr 流失的现象得到减缓。且Cr2+亦能被氧化成更高的价态,从而消耗氧化性物质。
对于BS-Cr3+,Cr3+能氧化合金的元素,且以铁、铬元素为主,从而明显加速腐蚀。
图9 和图10 分别为HX 在BS 以及BS-Cr3+熔盐中腐蚀100 h 后截面EDS 线扫及元素面分布图。由图可知,HX 分别在BS 和BS-Cr3+熔盐中腐蚀后元素分布表现出显著差异。图9 所示腐蚀层中O 和Mg元素比图10 的显著。这可能是BS-Cr3+熔盐中CrCl3消耗了部分熔盐中的溶解O2,使图10腐蚀层中形成的MgO 减少,但不妨碍CrCl3氧化合金中的Cr和Fe,促进富铬富铁合金腐蚀。图10显示HX腐蚀层中的铬和铁明显减少,说明熔盐中引入CrCl3会促进合金中铬和铁的流失。
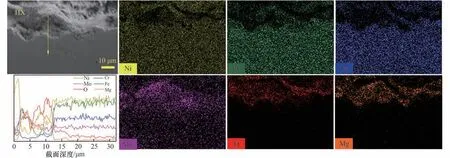
图9 HX在BS熔盐中腐蚀100 h后截面EDS线扫结果(沿黄线)及元素分布Fig.9 EDS line scanning results(along yellow line)and cross section element distribution of HX after corrosion in BS molten salt for 100 h

图10 HX在BS-Cr3+熔盐中腐蚀100 h后截面EDS线扫结果(沿黄线)及元素分布Fig.10 EDS line scanning results(along yellow line)and cross section element distribution of HX after corrosion in BS-Cr3+molten salt for 100 h
2.5 熔盐中铬价态对镍基金属腐蚀的热力学分析
当富铬合金浸入液态熔盐中时,盐中的氧化性杂质,如熔盐中溶解的O2、H2O 和水解产物HCl 以及由O2在腐蚀过程产生的Cl2等均会引起合金腐蚀[33]。为反映各种元素的流失倾向,计算了构成合金组成元素的标准摩尔生成Gibbs 自由能ΔfG⊖m,如表3 所示,相关物质参数采用HSC9数据库中的参数。

表3 合金元素的氯化物600℃下的标准摩尔生成Gibbs自由能Table 3 Molar Gibbs free energy of formation of metal chloride at 600℃
可见,Cr 的氯化物生成Gibbs 自由能最负,因而其还原性最高,最容易被优先氧化;其次是Fe。因而,在表2中显示Cr与Fe的优先流失。
当熔盐中引入Cr0,由于所添加的金属Cr 粉会优先消耗溶解于熔盐中的氧化性物种(如溶解O2),发生如式(4)、式(5)的反应(ΔrG⊖m为反应的标准Gibbs自由能)。
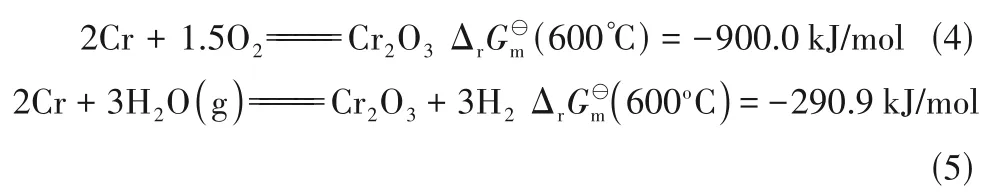
上述反应可降低熔盐中氧化性物种的量,这与添加活性金属Mg[34]抑制腐蚀机理较为一致,从而使金属表面被侵蚀最少。
当熔盐中存在一定浓度的Cr2+可一定程度抑制合金中Cr的阳极溶解,因为合金表面Cr的阳极溶解首先发生式(6)的反应。
从CrCl3氧化Cr 和Fe 在600℃下反应的标准Gibbs 自由能小于-40 kJ/mol(表征反应进行完全的关键数据)来看[35],Cr3+对合金表面Cr与Fe 的氧化进行得十分彻底。而相同温度下CrCl3氧化Ni、Mo 反应为-29.6 kJ/mol、-22.5 kJ/mol,反应程度均小于上述两个反应。因而,CrCl3对HB-2 合金的加速腐蚀程度较弱。
同时,Cr3+仍具有一定的还原性,Cr3+还可被熔盐中的O2等氧化性杂质氧化成更高价的化合物[7,36],对于低Cr、Fe 的HB-2 合金,在该温度下Cr3+对金属镍和钼的氧化程度可能有限,而Cr3+被氧化,消耗了熔盐中的氧化性物质,从而总体上反而抑制其对HB-2 的腐蚀。可见,Cr 和Fe 流失不仅与熔盐中氧化性物种(例如溶解O2)有关[7],还与熔盐中引入铬的价态有关。
3 结 论
采用静态浸没法研究了NaCl-MgCl2-CaCl2熔盐中引入Cr0、CrCl2和CrCl3后,对镍基合金HB-2、HC-276 和HX 腐蚀性的影响,通过热力学计算说明了CrCl3的引入对富铬合金腐蚀性增强的原因,研究获得如下结论。
(1)NaCl-MgCl2-CaCl2熔盐中引入低价态Cr 和CrCl2,能有效降低HB-2、HC-276 和HX 的腐蚀程度;引入Cr3+则会促进两种富铬合金的腐蚀,但能抑制低铬合金HB-2的腐蚀。
(2)进入熔盐的Cr0和CrCl2,主要通过消耗熔盐中溶解的氧化性物种(如O2、H2O 等)来抑制金属的腐蚀。
(3)进入熔盐的CrCl3,对合金中的单质Cr 和Fe有很强的氧化性,从而促进富铬富铁合金HC-276和HX 的腐蚀。同时CrCl3本身具有还原性,对合金中单质Ni的氧化性有限,从而可抑制贫铬贫铁的镍钼合金HB-2的腐蚀。
