RIP1/RIP3介导的程序性坏死在LPS诱导的多巴胺神经元损伤中的作用
2022-08-09郑天成孙宪昌
郑天成 郭 俊 孙宪昌
1.山东第一医科大学(山东省医学科学院)临床与基础医学院,山东 泰安 271000;2.泰安市中心医院生殖医学科,山东 泰安 271000
帕金森病(Parkinson′s disease,PD)是一种多发于老年或老年前期的中枢神经系统原发性退行性疾病,其主要的临床表现有静止性震颤、肌肉僵直和姿势障碍等运动症状,部分患者伴有嗅觉功能减退、精神异常及认知功能障碍等非运动症状[1-2]。病理学研究已证实,PD的发病主要与中脑黑质致密部多巴胺能神经元进行性变性缺失及纹状体内多巴胺递质的耗竭有关。因此,了解多巴胺神经元死亡的原因和途径是干预、治疗PD的关键,但多巴胺神经元变性缺失的具体机制至今仍未完全清楚;目前尚无有效的方法或药物能逆转或阻断PD病情的进展。
研究显示,PD 的发病机制十分复杂,可能与环境、年龄、神经炎症和遗传等多种因素密切相关,在以上多种因素的共同作用下最终导致了中脑黑质内多巴胺能神经元的选择性变性、缺失直至死亡[3-4]。由于黑质致密带内多巴胺能神经元的死亡是不可逆的,因此预防或治疗PD 的关键在于病因预防和对多巴胺能神经元死亡机制的干预。关于多巴胺能神经元死亡机制和形式目前仍存争议,大部分研究认为,黑质内多巴胺能神经元的死亡主要与 caspase 依赖的凋亡有关[5-6],多巴胺能神经元在不同刺激因素的作用下,通过死亡受体或内质网途径激活细胞内caspase级联反应,诱导多巴胺能神经元以凋亡形式死亡缺失。但有研究发现,应用凋亡抑制剂并不能完全阻断多巴胺能神经元的死亡,说明其他死亡形式也参与了多巴胺能神经元的损伤过程。进一步研究发现,程序性死亡(necroptosis)可能参与了多巴胺能神经元的死亡并在其中发挥着重要作用[7]。
程序性死亡是近年来发现并被证实的一种主要由肿瘤坏死因子受体家族或细胞表面Toll样受体家族触发启动,可受严格调控的非caspase依赖性的新型细胞死亡形式。大量研究已证实,程序性死亡与免疫性疾病、炎症、中枢神经系统退行性疾病以及肿瘤等多种人类疾病的发生发展密切相关[8];但目前关于程序性死亡与PD的关系研究相对较少且存在争议。本研究利用脂多糖(lipopolysaccharide,LPS)建立的PD 动物模型,探讨了程序性死亡在中枢神经系统炎症反应和多巴胺能神经变性缺失中的作用。
1 材料与方法
1.1 实验动物
本实验选用的动物为30 只2~3 月龄、体质量200~250 g、雌雄不限的成年SD 大鼠,购自山东省实验动物中心(实验动物许可证号:SYXK(Lu)20190022)。大鼠适应环境7 d后开始实验,实验过程中将大鼠置于室温(21±2)°C、昼夜循环光照、能自由进食和饮水条件下饲养。本实验经山东第一医科大学动物伦理委员会批准。
1.2 药品与试剂
阿扑吗啡(apomorphine,APO)购自美国Sigma公司;LPS 与 Nec-1 购自美国 MCE 公司,LPS 使用前用生理盐水(normal saline,NS)溶解成2.5 µg/µL 的备用液,Nec-1用NS(含10%PMSF,20%SBE-β-CD)溶解成1 mg/mL的备用液;水合氯醛购自上海易恩化学技术有限公司;多克隆小鼠抗大鼠-TH抗体购自美国Sigma 公司;小鼠抗-CD11b 单克隆抗体购自美国Millipore公司;免疫组化试剂盒(SABC法)购自北京中杉金桥生物技术有限公司;ELISA试剂盒、蛋白浓度测定BCA试剂盒及RIPA蛋白裂解液均购自上海碧云天生物技术有限公司;兔抗鼠RIP-1 及兔抗鼠RIP-3多克隆抗体均购自英国Abcam公司;多巴胺、双羟苯乙酸及高香草酸标准品购自美国Sigma公司;其它化学试剂购自国内商业公司,均为分析纯。
1.3 主要仪器
OLYMPUS BX50型正置显微镜,日本OLYMPUS公司;专用大鼠立体定位仪,中国深圳瑞沃德生命科技有限公司;CM-2000型切片机(冰冻),德国莱卡公司;多功能酶标分析仪,美国Molecular Devices(MD)公司;高效液相色谱仪及2465型电化学检测器,美国Waters公司;UVP Biospectrum,美国UVP公司。
1.4 实验方法
1.4.1 实验分组及给药 SD 大鼠随机分为3 组:(1)LPS 模型组(n= 10),动物经水合氯醛(腹腔注射,400 mg/kg)麻醉后,沿颅顶中线切开头部皮肤,棉签蘸取30%H2O2氧化腐蚀皮下组织暴露颅骨,以颅骨骨缝表面前、后囟为标志,参考大鼠脑立体定位图谱,确定大鼠右侧黑质注射坐标为门齿杆−2.3 mm;前囟后−5.2 mm;旁开 2.1 mm;深度−7.8 mm。用微量注射器吸取2 µL(5 µg)LPS 以1 µL/min 的速度注射到大鼠右侧黑质区[9],给药完毕留针5 min后缓慢退出,骨蜡封闭颅骨表面钻孔,最后缝合头部皮肤。LPS 注射后,腹腔内连续注射含 10% PMSF 和 20% SBE-β-CD 的 NS(2 mL/kg)14 d。(2)Nec-1+LPS 组(n=10),动物黑质内注射2 µL LPS(方法同模型组),然后腹腔内注射Nec-1(2 mg/kg),连续用药14 d。(3)对照组(n= 10),黑质内注射NS 2µL,其余用药同LPS模型组。
1.4.2 行为学检测 黑质内注射LPS 14 d后,所有动物进行行为学检测。检测时提前15 min 将动物放置于旋转记录仪中使其适应测试环境,然后皮下注射 APO 0.5 mg/kg(用 NS 溶解成 1 mg/mL 的备用液),再将大鼠放置于旋转记录仪中观察其行为学改变,记录30 min内大鼠由未注射侧向注射侧旋转的总圈数进行统计学分析。
1.4.3 高效液相色谱-电化学(high performance liquid chromatography,HPLC)检测 旋转行为检测后,各组取6只动物,麻醉后断头处死并立即分离出两侧纹状体,快速称重后冻存于−80ºC 备用。实验时取出冻存有纹状体的EP 管,加入预冷的样品预处理A 液(0.4 M HClO4)300 µL,用电动匀浆机匀浆,然后4 ºC,13 000 r/min 离心30 min;离心后吸取240µL上清液存放于1.5 mL EP管,加入120µL预冷的预处理B液(300 mM磷酸氢二钾,2 mM EDTA·2Na,20 mM 柠檬酸钾),4 ºC,13 000 r/min 离心30 min 后吸取上清液上机检测。色谱柱为十八烷基硅烷分析柱,检测方法严格按照说明进行,最大上样柱压 2 700 psi,流速 1.0 mL/min,ECD 电压0.65 V,抽样体积20 µL,每一样品检测时间为50 min。根据说明利用多巴胺(dopamine,DA)、高香草酸(homovanillic acid,HVA)及二羟苯乙酸(dihydroxyphenylacetic acid,DOPAC)标准品的检测结果计算样品中DA、HVA和DOPAC的含量。
1.4.4 免疫组化 每组选取4 只大鼠,麻醉后用4%多聚甲醛经升主动脉灌注固定脑组织。灌注结束后剥离出全脑,放置于4%多聚甲醛中后固定12 h,然后将固定好的脑组织依次放入10%、20%及30%的蔗糖溶液中进行梯度脱水。标本处理好后用OCT包埋剂包埋,然后根据大鼠脑定位图谱确定黑质的位置为前囟后4.8 mm 至6.0 mm,利用冰冻切片机在此范围内进行连续冠状切片,片厚30µm;切片被间隔分成2套,分别进行CD11b及TH免疫组化染色。染色采用自由漂片法进行,过程简述如下:切片用4%胎牛血清常温孵育20 min 封闭非特异性抗原,分别加入一抗(TH 1∶800,CD11b 1∶250)4 ℃摇床过夜,然后加入生物素化的二抗(1∶1 200),常温孵育1 h,再加入被辣根过氧化物酶标记过的三抗(1∶1 000),常温孵育1.5 h,最后应用DAB进行显色。
利用OLYMPUS FV1000 Viewer 软件对脑片黑质区内TH 反应阳性的细胞计数,每侧黑质计数两遍,取其平均数作为该侧TH反应阳性细胞的数目,然后用损伤侧的细胞数与未损伤侧的细胞数进行对比,即为损伤侧黑质TH神经元存活率[9]。
1.4.5 酶联免疫吸附实验(enzyme-linked immunosorbent assay,ELISA) 行为学检测结束后,每组随机选取3 只大鼠,经水合氯醛麻醉后断头取脑,快速切去小脑及嗅球部位,在大脑中后1/3交界处垂直切开脑组织,在大脑的冠状切面可见清晰的黑质轮廓,用弯头眼科镊迅速夹取两侧黑质部位的脑组织,称重后−80ºC保存备用。ELISA实验前,将组织样品按1∶9 的比例加入预冷的lysis 缓冲液(含有蛋白酶抑制剂)冰上匀浆,4 ℃,13 000 r/min 离心30 min 后吸取上清液,ELISA 检测黑质中 TNF-α 及IL-1β 表达量的变化,检测方法严格按照试剂盒说明进行。根据样本450 nm 处测得的吸光度(optical density,OD)对结果进行分析,以标准品浓度作横坐标,OD值为纵坐标,绘制出标准曲线并算出回归方程。最后根据待检样本的OD值即可得到相应的蛋白含量,乘以稀释倍数后再与样品蛋白浓度作比较,即为该样品的最终浓度。
1.4.6 免疫印迹实验 每组选取3 只大鼠用于免疫印迹实验,根据1.4.5 的方法取大鼠黑质组织并称重后保存于−80℃冰箱中备用。实验时每个样品按照每4 mg 100 uL 裂解液的比例加入含有1 mM PMSF的RIPA裂解液,用电动匀浆机在冰上将组织充分匀浆裂解,抽提组织中的蛋白。应用BCA法检测各样品的蛋白浓度,从每个样品中吸取等量蛋白(30µg)进行聚丙烯酰胺凝胶(10%的分离胶)电泳以分离样品中的不同蛋白,然后用湿式电泳转移的方法将凝胶中的蛋白转移到PVDF 膜上。PVDF 膜用5%脱脂牛奶封闭过夜,然后将膜移入含有一抗CD11b(1∶400),RIP1(1∶1 000),RIP3(1∶1 000)和 β-actin(1∶5 000)的孵育盒中4 ℃摇床过夜;再用辣根过氧化酶标记的二抗(1∶8 000)37 ℃孵育1 h,膜上滴加ECL化学发光液进行显色。应用自动化学发光成像分析系统显影拍照并进行积分光密度(integrated optical density,IOD)测定,目的蛋白与β-actin的IOD比值用于统计分析。
1.5 统计学处理
实验所得结果用均数±标准差()表示,应用GraphPad Prism 6.0 统计学软件对数据进行单因素方差分析,采用Newman-Keuls 法进行两两比较。检验水准α=0.05。
2 结 果
2.1 大鼠旋转行为的改变
研究证实,LPS 单侧黑质区注射会损伤患侧黑质-纹状体通路,导致同侧纹状体内DA递质含量显著减少,是建立炎症相关PD 动物模型的较好方法[10-11],而APO 诱导的动物旋转行为可以作为衡量此类PD 动物模型是否成功建立的良好指标[12-13]。本实验在LPS 注射14 d 后检测了APO 诱导的大鼠行为学改变,结果发现,对照组大鼠在注射APO 后并没有出现明显的旋转现象;LPS 模型组大鼠在皮下注射APO后出现明显的旋转行为,旋转时大鼠以患侧后肢作为支点,身体向患侧弯曲,首尾相衔接,原地转动,有的动物甚至出现身体翻转现象,30 min内旋转的平均速度达8.5 r/min,说明注射侧黑质多巴胺神经元有明显损伤缺失,已达到PD成模标准;而应用Nec-1 干预后,大鼠的旋转行为较LPS 组明显改善(图1)。
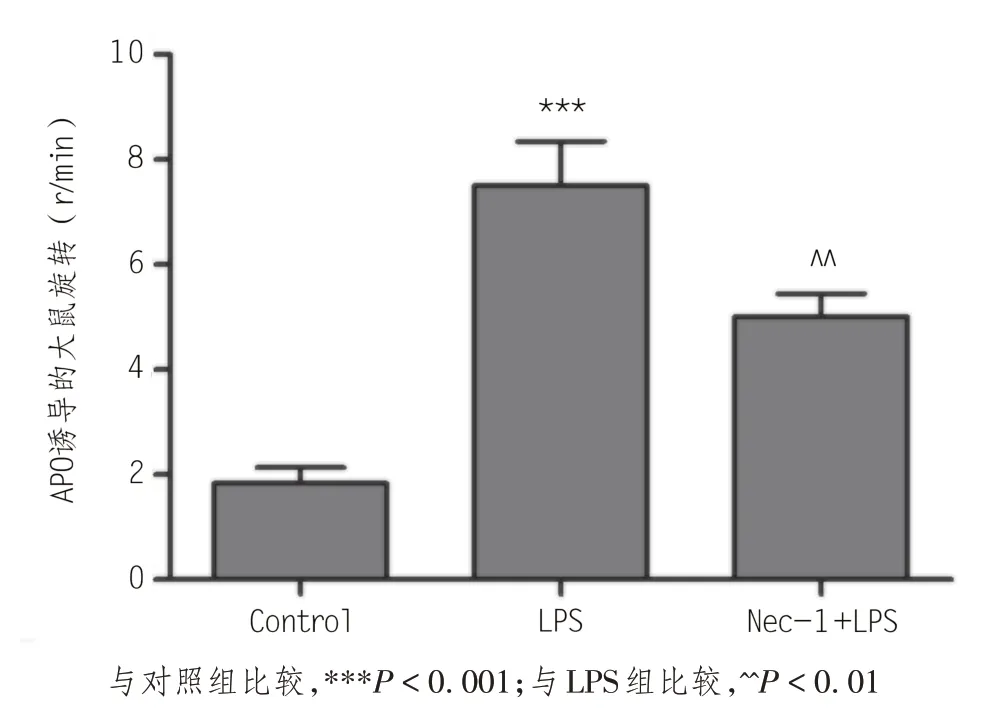
图1 阿扑吗啡诱导的旋转行为
2.2 大鼠纹状体内多巴胺及其主要代谢产物含量的变化
纹状体内DA 递质的大量丢失是导致PD 患者出现运动症状的主要原因,与黑质内多巴胺能神经元的受损缺失程度密切相关。为进一步明确LPS诱导PD 的机制及Nec-1 对多巴胺能神经元损伤的影响,本研究应用HPLC法检测了大鼠纹状体内DA及主要代谢产物DOPAC和HVA含量的变化。结果如图2所示,LPS模型组大鼠患侧纹状体内DA及其代谢产物HVA 和DOPAC 的含量较对照组明显减少;Nec-1+LPS 组大鼠患侧纹状体内DA、HVA 及DOPAC 的含量与LPS 组相比,均有不同程度增高,差异均有统计学意义(P<0.05)。

图2 纹状体内多巴胺及其代谢产物含量的变化
2.3 大鼠黑质内酪氨酸羟化酶(tyrosinehydroxylase,TH)阳性神经元的形态及数量变化
TH是多巴胺能神经元内特有的酶系统,利用免疫组织化学的方法对其特异性染色后可以直观的反应黑质内多巴胺神经元的形态及数量变化,从而能更好地观察PD的病理变化。本实验免疫组织化学染色结果发现,对照组黑质内多巴胺能神经元被染成棕黄色,胞体较大,境界清楚,纤维丰富,没有明显的损伤和缺失;LPS组大鼠患侧黑质内TH染色明显变浅,阳性神经元数量较健侧明显减少,TH阳性神经元存活率仅有31.6%;Nec-1+LPS组大鼠损伤侧黑质区TH免疫阳性神经元数量较未损伤侧也有显著下降,但与LPS 组动物的损伤侧比较,其TH 免疫阳性神经元数量有明显提升,其存活率上升到51.9%,差异有统计学意义(P<0.05)。各组健侧黑质内TH染色情况差异无统计学意义(P>0.05)。见图3。
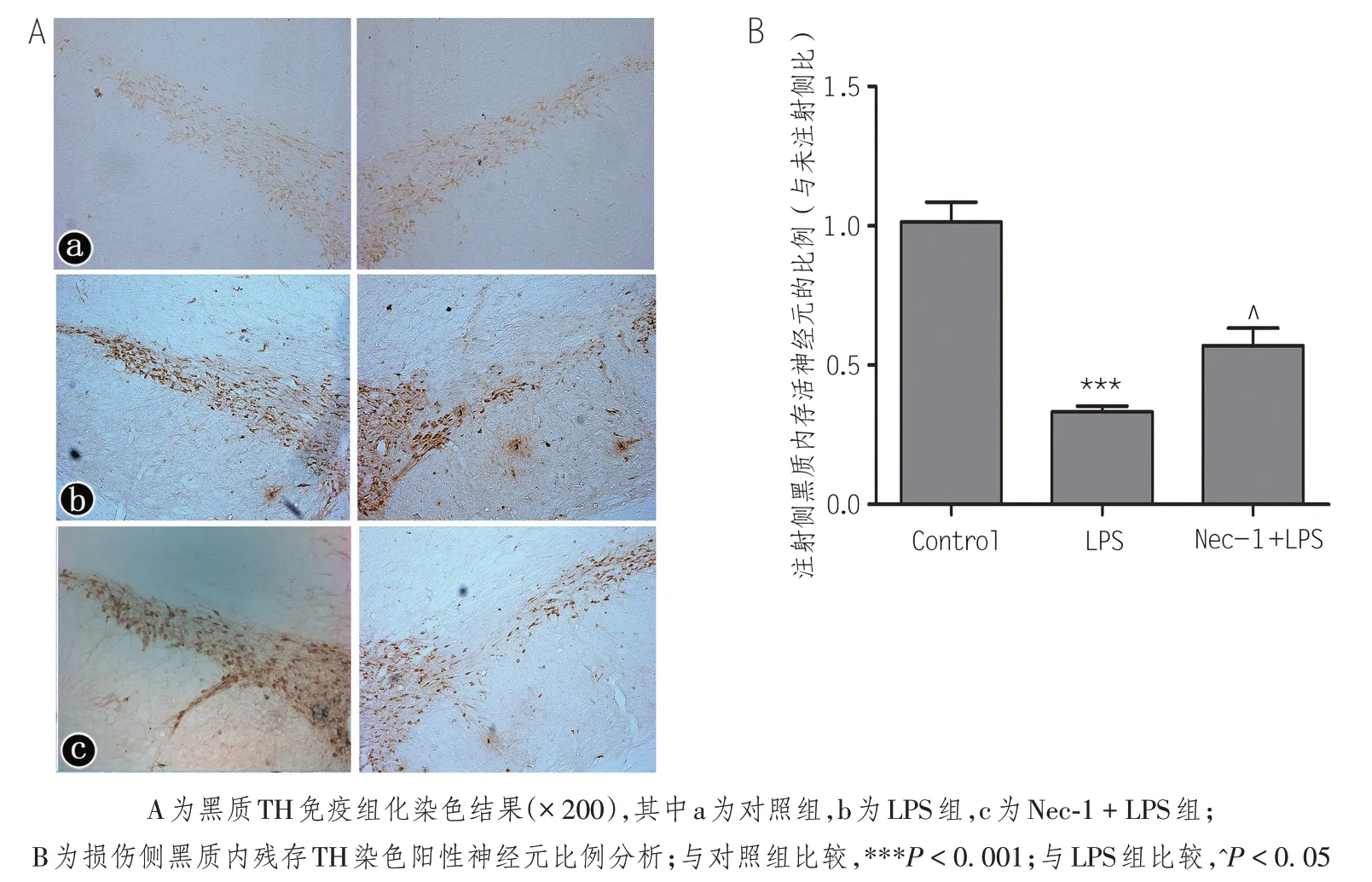
图3 黑质内TH免疫阳性神经元数量变化
2.4 大鼠黑质内小胶质细胞的激活及Nec-1 的影响
LPS诱导的以小胶质细胞激活为特征的中枢神经炎症是导致黑质多巴胺能神经元损伤、变性甚至死亡缺失的重要原因。为了观察中枢神经系统炎症反应在PD致病过程中的作用及程序性坏死对神经炎症的影响,本实验应用免疫组化的方法对小胶质细胞特异表达的蛋白CD11b进行免疫染色,观察各组动物黑质内小胶质细胞的形态及数量变化并以此判断其活化情况和中枢炎症反应的程度。结果如图4所示,对照组黑质区内可见胞体较小、形态呈明显分枝状、散在分布的静息小胶质细胞;LPS组动物损伤侧黑质内小胶质细胞数量明显增多、胞体显著增大、细胞分枝消失、形态呈阿米巴样变化,是典型的小胶质细胞过度激活的反应,免疫印迹结果显示,CD11b 的表达量明显较对照组增加;Nec-1+LPS组大鼠损伤侧黑质内小胶质细胞也有激活的反应,表现为数量增多、染色程度较深、阿米巴样细胞增多,但与LPS组相比范围较局限、活化细胞数量较少,染色吸光度及CD11b的表达量明显下降,与LPS组相比差异有统计学意义(P<0.05),说明Nec-1对LPS诱导的炎症有抑制作用。

图4 损伤侧黑质内CD11b免疫组化及免疫印迹结果
2.5 大鼠黑质炎症因子 TNF-α 和 IL-1β 含量的变化
小胶质细胞活化后分泌的促炎介质TNF-α 和IL-1β 与DA 能神经元的进行性变性死亡密切相关。为了进一步探讨LPS 诱导的黑质多巴胺能神经元的死亡机制,本实验利用ELISA 方法检测了大鼠黑质区主要炎症因子 TNF-α 及 IL-1β 的含量变化。结果如图5 所示,与对照组相比,LPS 组动物损伤侧黑质内TNF-α 和IL-1β 的含量明显升高;与LPS 组相比,注射LPS 后应用Nec-1 进行干预的动物,其损伤侧黑质区TNF-α 及 IL-1β 的含量明显降低。以上结果说明,LPS 激活小胶质细胞后释放的炎症因子是诱导多巴胺能神经元死亡的重要原因,炎症反应及炎症因子的增加可能参与了细胞程序性坏死的过程。
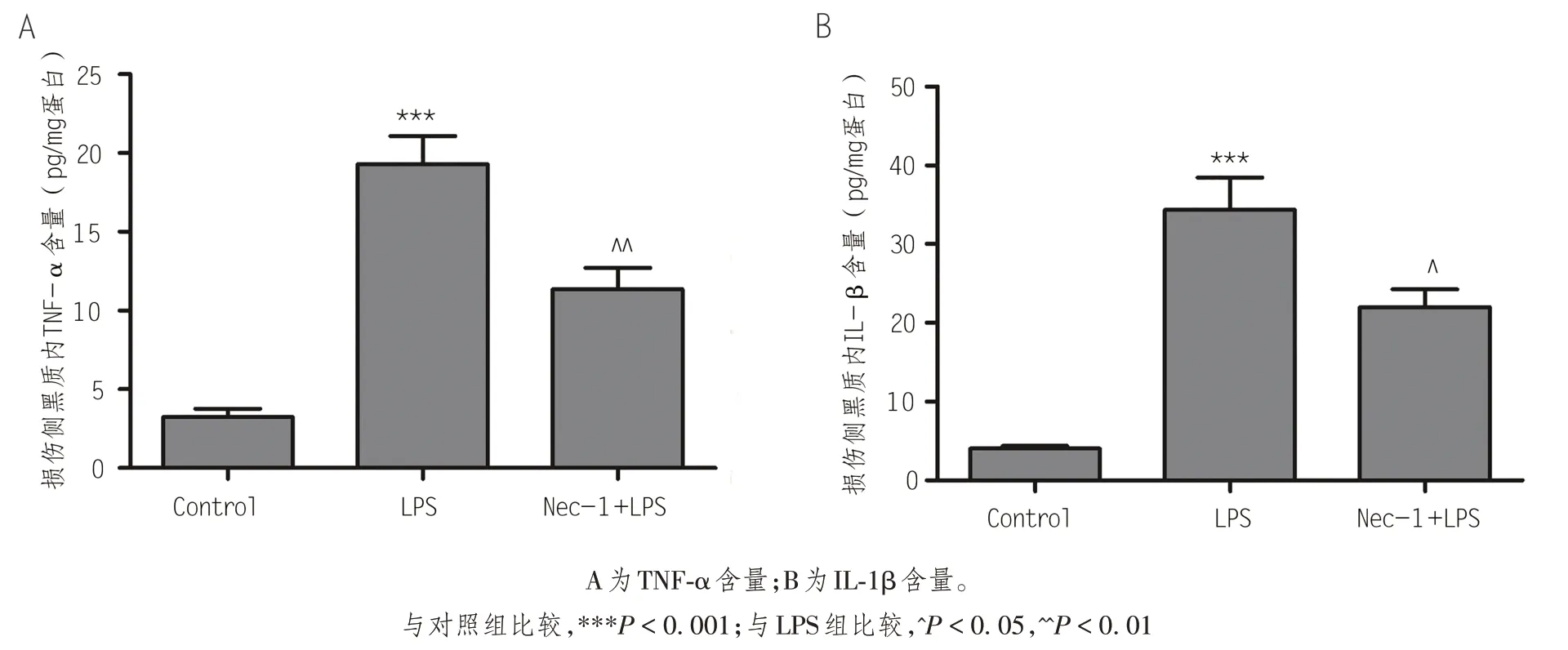
图5 损伤侧黑质内炎症因子表达量变化
2.6 大鼠黑质内程序性死亡相关蛋白RIP1 与RIP3的表达量变化
RIP1与RIP3是细胞启动程序性坏死的关键信号分子,是反映细胞死亡形式的重要蛋白。为了进一步探讨程序性死亡与LPS诱导的多巴胺能神经元死亡的关系及PD 的发病机制,本实验应用免疫印迹的方法检测了大鼠损伤侧黑质内RIP1与RIP3蛋白表达量的变化。结果如图6 所示,LPS 组大鼠损伤侧黑质内RIP1与RIP3蛋白的表达量较对照组均有明显增加;而Nec-1+LPS 组大鼠损伤侧黑质内RIP1与RIP3蛋白的表达量较LPS组均显著降低,差异均有统计学意义(P<0.05)。
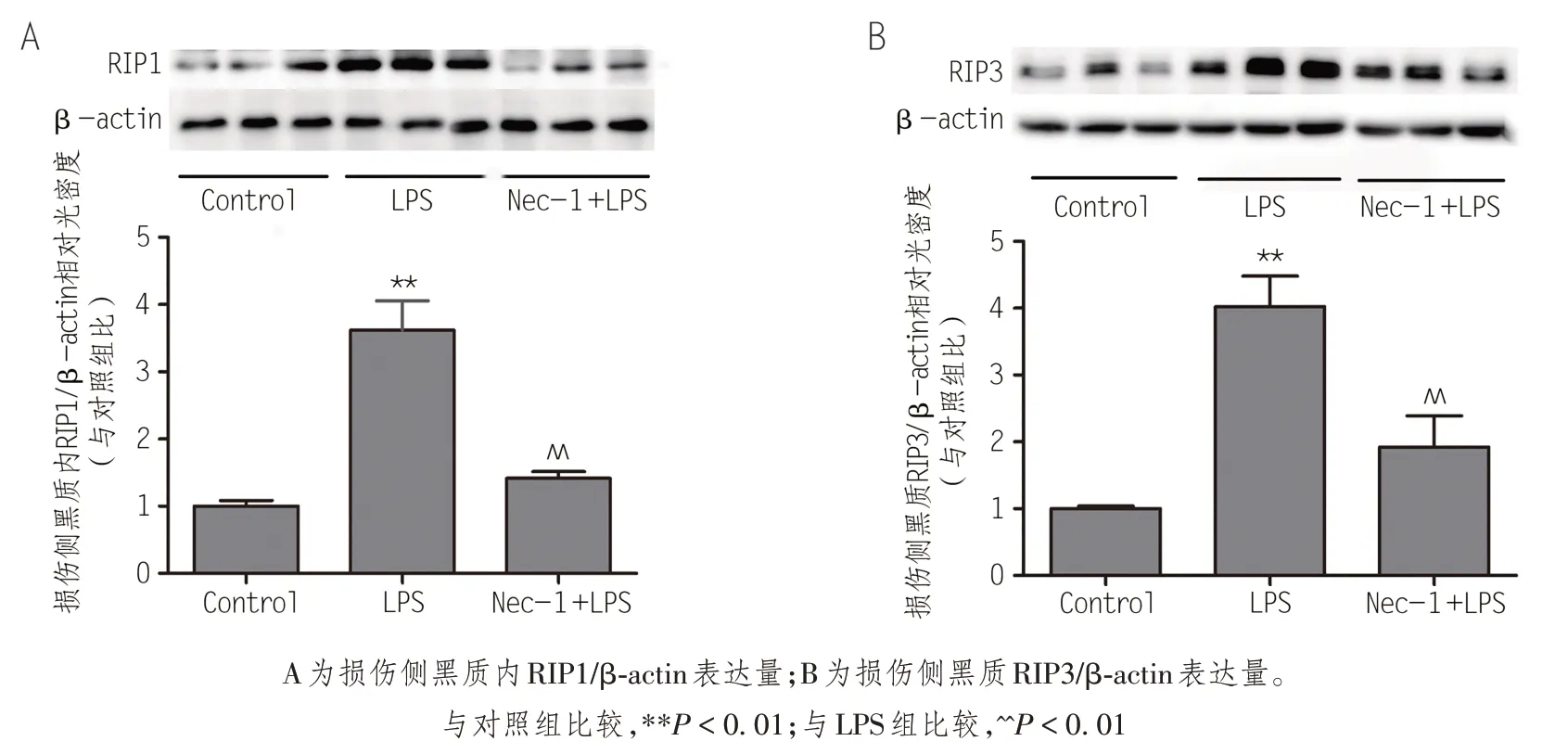
图6 损伤侧黑质内程序性死亡相关蛋白表达量变化
3 讨 论
PD 是一种多发于中老年人群的神经退行性疾病,是世界第一大运动障碍性疾病,60 岁以上人群中患病率为1%~4%,目前全球PD患者总人数已达600万人,其中我国患者约占全球的一半[14],而且随着我国人口老龄化的加重,PD患者的数量在未来十年内将会明显增加,PD已成为不可小觑的公共卫生问题。PD 主要的临床表现为静止性震颤、肌肉僵硬、运动迟缓和姿势不稳等运动功能障碍,尽管这在短期内不会危及患者生命,但其较高的发病率、致残率及较长的病程会严重影响患者的生活质量并给社会带来沉重的经济负担,因此关于PD 的研究一直是国内外神经科学领域的研究重点。
研究已证实,中脑黑质致密部多巴胺能神经元进行性变性缺失及纹状体内多巴胺递质的大量耗竭是PD的主要病理改变,但PD的确切病因及发病机制迄今未明,多数学者认为PD 患者多巴胺神经元的死亡可能与遗传、环境、年龄及神经炎症等多种因素相关,是多种因素共同作用的结果[15]。越来越多的研究发现,以小胶质细胞激活为特征的中枢神经炎症在PD 的发病过程中起着关键性作用[16]。小胶质细胞是脑内固有的免疫监视细胞,对脑内各种炎症刺激反应灵敏,其适度激活可清除脑内病原、细胞碎片,有助于维持大脑内环境稳态;但持续或较强的炎症刺激会导致脑内小胶质细胞过渡激活并释放谷氨酸、促炎因子或活性氧等大量的有害物质损伤神经元,神经元损伤裂解后可以释放出大量的促炎因子又进一步加剧中枢神经炎症反应,这样会在脑内形成一个持续存在的炎症反应与神经元损伤的恶性循环,最终导致了多巴胺能神经元的大量损伤、死亡及PD 的发生。中枢神经炎症被认为是诱发PD 的一个独立危险因素,开始受到越来越多研究者的关注;炎症PD 模型随之成为研究PD机制、探索PD治疗方法的常用模型。因此,本实验应用黑质内单次注射LPS 的方法建立炎症PD 动物模型进行了相关研究。实验结果与文献报道基本一致[9,17],黑质内注射LPS可成功诱导脑内炎症反应以及黑质内多巴胺能神经元的大量死亡缺失,HPLC检测发现损伤侧纹状体内多巴胺递质明显减少,APO皮下注射可诱导出模型组动物明显的旋转行为,说明实验成功复制了炎症相关PD大鼠模型。本实验以此动物模型为基础,进一步对PD 多巴胺能神经元死亡方式和机制进行了研究。
众所周知,多巴胺能神经元丢失后是不可再生的,因此预防或治疗PD 的关键在于病因预防和死亡机制的干预,只有尽可能的减少或延缓多巴胺能神经元的损伤和死亡,才能有效地延缓或暂停PD的发生或进展。明确多巴胺能神经元的死亡方式和机制是治疗和干预PD至关重要的环节,目前关于多巴胺能神经元死亡的机制和形式仍未完全清楚,大部分研究认为黑质内多巴胺能神经元的死亡主要与caspase依赖的凋亡有关,多巴胺能神经元是在不同刺激因素的作用下,通过死亡受体或内质网途径激活胞内caspase级联反应,最终诱导了多巴胺能神经元以凋亡的形式变性死亡[5-6]。但进一步研究发现,应用凋亡抑制剂并不能完全阻断多巴胺能神经元的死亡,说明其它细胞死亡形式也参与其中。最新的研究指出,程序性细胞坏死可能参与了多巴胺能神经元的死亡并在其中发挥着重要作用[7,18]。
程序性死亡是近年来发现并被证实的一种可调控的、caspase 非依赖性的细胞死亡形式,主要由肿瘤坏死因子受体家族及细胞表面Toll样受体家族触发启动[19],然后依次磷酸化激活细胞内重要信号分子RIP1和RIP3并形成坏死复合物,最后激活混合谱系激酶域样蛋白(mixed lineage kinase domain-like protein,MLKL)使其形成寡聚体并转移到质膜上,促进离子内流(钙和钠)或孔隙形成,导致细胞能量代谢障碍并使膜完整性受到破坏,触发程序性细胞坏死并释放出具有损伤作用的分子引起加重炎症反应,参与不同的生理病理过程[20]。目前关于程序性细胞坏死与PD的研究相对较少,但有证据表明,程序性细胞坏死在PD的病理变化中发挥重要作用,可以为PD 的治疗提供潜在靶点。Lin 等[21]研究发现,在MPTP制备的小鼠PD模型中,程序性坏死相关的重要蛋白RIP1、RIP3、MLKL 表达量明显增加;应用RIP1阻断剂Necrostatin-1(Nec-1)阻断程序性坏死后可使上述蛋白表达量明显下降,同时明显减少了DA神经元的损伤数量;另外该研究还发现,敲除了RIP3和MLKL 的小鼠除程序性坏死受到抑制外,中脑黑质内的促炎因子表达量也有明显降低。Onate 等[22]在最近的研究中也得到了类似的结果,证实了程序性坏死参与了6-OHDA诱导的DA神经元死亡,Nec-1处理或抑制RIP3的表达可保护多巴胺能神经元。但Dionísio等[23]研究发现,凋亡依然是MPTP诱导的多巴胺能神经元死亡的主要方式,程序性细胞坏死在其中的作用有限。所以关于程序性细胞坏死在PD中的作用机制仍然存在争议且研究较少。因此,本实验通过LPS建立炎症诱导的PD模型,利用程序性坏死特异阻断剂Nec-1观察了程序性坏死在炎症诱导的多巴胺能神经元死亡中的作用。结果发现,Nec-1能明显改善LPS诱导的动物旋转行为,明显减轻黑质内多巴胺能神经元的损伤和丢失,明显提升了纹状体内多巴胺递质的含量,表明程序性坏死是LPS导致多巴胺能神经元丢失的重要方式。本实验还发现,作为脑内炎症反应重要标志的小胶质细胞,在LPS刺激下明显激活,表现为数量明显增多、胞体明显变大、突起变短甚至消失,呈现阿米巴样形态;而应用Nec-1后小胶质细胞数量、形态学变化均有明显改善。以上结果表明,抑制程序性细胞坏死可通过减轻炎症反应保护多巴胺能神经元。这些结果与既往研究基本一致,均表明炎症反应可诱导程序性坏死的发生,程序性坏死可释放促炎物质加剧脑内炎症反应的进行,程序性坏死可作为治疗神经系统炎症疾病的重要靶点,值得进一步研究。
综上所述,本研究通过LPS 建立的炎症PD 模型,证实了程序性坏死与多巴胺能神经元的死亡密切相关,可能参与了PD 的发病过程。而程序性坏死具体的机制,在多巴胺能神经元死亡中的重要程度,凋亡和程序性坏死之间的关系等诸多问题,仍需要通过进一步的实验去探讨。
利益冲突所有作者均声明不存在利益冲突
