Dying by fire: noncanonical functions of autophagy proteins in neuroinflammation and neurodegeneration
2022-08-08AlexisRickmanAddisonHilyardBradleeHeckmann
Alexis D. Rickman,Addison Hilyard,Bradlee L. Heckmann
Abstract Neuroinflammation and neurodegeneration are key components in the establishment and progression of neurodegenerative diseases including Alzheimer’s Disease (AD). Over the past decade increasing evidence is emerging for the use of components of the canonical autophagy machinery in pathways that are characterized by LC3 lipidation yet are distinct from traditional macro-autophagy. One such pathway that utilizes components of the autophagy machinery to target LC3 to endosomes,a process termed LC3-associated endocytosis (LANDO),has recently been identified and regulates neuroinflammation. Abrogation of LANDO in microglia cells results in a propensity for elevated neuroinflammatory cytokine production. Using the well-established 5xFAD model of AD to interrogate neuroinflammatory regulation,impairment of LANDO through deletion of a key upstream regulator Rubicon or other downstream autophagy components,exacerbated disease onset and severity,while deletion of microglial autophagy alone had no measurable effect. Mice presented with robust deposition of the neurotoxic AD protein β-amyloid (Aβ),microglial activation and inflammatory cytokine production,tau phosphorylation,and aggressive neurodegeneration culminating in severe memory impairment. LANDO-deficiency impaired recycling of receptors that recognize Aβ,including TLR4 and TREM2. LANDO-deficiency alone through deletion of the WD-domain of the autophagy protein ATG16L,revealed a role for LANDO in the spontaneous establishment of age-associated AD. LANDO-deficient mice aged to 2 years presented with advanced ADlike disease and pathology correlative to that observed in human AD patients. Together,these studies illustrate an important role for microglial LANDO in regulating CNS immune activation and protection against neurodegeneration. New evidence is emerging that demonstrates a putative linkage between pathways such as LANDO and cell death regulation via apoptosis and possibly necroptosis. Herein,we provide a review of the use of the autophagy machinery in non-canonical mechanisms that alter immune regulation and could have significant impact in furthering our understanding of not only CNS diseases like AD,but likely beyond.
Key Words: aging; Alzheimer’s disease; autophagy; inflammation; LC3-associated endocytosis; microglia; neurodegeneration; neuroinflammation
Introduction
Macro-autophagy (autophagy henceforth) has been shown to be a contributing pathway to the regulation of immune responses and inflammation with primary roles in modulating metabolic and cellular homeostasis. In this canonical form of autophagy,double-membraned vacuoles known as autophagosomes are responsible for the collection and deliverance of various intracellular materials to the lysosome for degradation as a response to stressors,such as starvation and nutrient deprivation (Dikic and Elazar,2018). Additionally,autophagy has been shown to participate in the regulation of various immune pathways,including in the regulation of the type-I interferon response,which in turn helps combat viral infections more effectively (Martin et al.,2018; Jin,2019; Tian et al.,2019). Likewise,autophagy has been implicated in regulating pro-inflammatory cytokine signaling including interleukin (IL)-1β secretion by targeting the IL-1β precursor,pro-IL-1β for degradation (Zhang et al.,2015; Claude-Taupin et al.,2018; Iula et al.,2018). Much investigation has led to the identification of multiple regulatory genes and proteins that govern the orchestrated processing and conjugation of the microtubule-associated protein light-chain 3 (LC3) to phosphatidylethanolamine residues within the developing autophagosome membrane following autophagic activation,newer evidence is revealing distinct roles for the autophagy machinery in alternate pathways including LC3-associated phagocytosis (LAP) (Kim et al.,2013; Martinez et al.,2015; Martinez et al.,2016; Heckmann et al.,2017; Heckmann and Green,2019),and LC3-associated endocytosis (LANDO) (Heckmann et al.,2019; Birgisdottir and Johansen,2020). We often described these,and similar pathways as “non-canonical functions of the autophagy machinery”.
Search Strategy and Selection Criteria
Studies cited in this review published from 2010 to 2020 were searched on the PudMed database using the following keywords: autophagy,neuroinflammation,Alzheimer’s disease,neurodegeneration,neuronal cell death,LC3-associated endocytosis,LC3-associated phagocytosis,LAP,LANDO,inflammation,beta-amyloid,inflammasome,NLRP3,Parkinson’s disease,amyotrophic lateral sclerosis,Huntington’s disease,CNS pathologies,IL-1beta,TNF-alpha,tau pathology.
Role of the Autophagy Machinery outside of Autophagy
Interestingly,although quite similar at a genetic level; autophagy,LAP,and LANDO are distinct cellular entities. A large share of the machinery found in the canonical autophagy pathway is needed for both LAP and LANDO,however components of the autophagy initiation complex including FIP200 and ULK1 are dispensable for LAP and LANDO,whereas the run-domain containing protein Rubicon,long held as an autophagic inhibitor is obligatory for LAP and LANDO and as shown previously is expendable for autophagy (Martinez et al.,2015). In the context of the central nervous system (CNS),canonical autophagy has been shown to play significant roles in both neuronal development and homeostatic maintenance in adult organisms (Sumpter and Levine,2011; Andres-Alonso et al.,2020; Fleming and Rubinsztein,2020; Kuijpers et al.,2020). Moreover,autophagic dysregulation in neurons has been identified in a number of CNS diseases including Huntington’s and Amyotrophic Lateral Sclerosis. Autophagic induction has therefore been proposed as a putative therapeutic avenue in these and other diseases of the brain (Cheon et al.,2019; Djajadikerta et al.,2020).
Similar to autophagy in neurons,the non-canonical functions of the autophagy machinery in LANDO have been shown to be key in preventing exacerbated β-amyloid accumulation and in mitigating β-amyloid induced neuroinflammation in a model of Alzheimer’s disease (AD). AD is the leading form of dementia globally and is one of the most prevalent neurodegenerative disorders without viable therapy. β-Amyloid deposition is one of the earliest hallmark features of AD in humans and has long been thought to be the major driver of disease pathology (Murphy and LeVine,2010). Over the past decade it has been well demonstrated that a primary component of AD pathology is robust and pervasive neuroinflammation. In particular,inflammatory cytokines including IL-1β and tumor necrosis factor alpha (TNFα) have been shown to be elevated in the brains of AD patients compared to healthy age-matched counterparts (Kinney et al.,2018). In the brain,cytokines such as IL-1β and TNFα are predominately produced by the resident innate macrophage-like immune cell,the microglia. In addition to localized cytokine production by microglia,peripheral cytokines have been shown to contribute in AD,especially as permeability of the blood-brain barrier increases concurrent with disease progression (Kinney et al.,2018).
Noncanonical Uses of the Autophagy Machinery in Neurodegeneration
In a previous study,we identified a role for LANDO in the recognition and clearance of β-amyloid in a murine AD model (Heckmann et al.,2020a). We found that abrogation of LANDO in microglia,through genetic deletion of Rubicon led to an increase in extracellular β-amyloid deposition,a robust increase in microglial activation and neuroinflammation,as well as exacerbated tau pathology and neurodegeneration.In vitroanalysis in cultured microglia clearly revealed that LANDO-deficiency alters the activation and inflammatory cytokine production following exposure to neurotoxic β-amyloid.In vivostudies were performed in mice on the 5xFAD background,a humanized transgenic model that leads to β-amyloid pathology (Heckmann et al.,2020a). Abrogation of LANDO greatly worsened the severity of disease and hastened the onset of pathology. Interestingly,we found that inhibition of canonical autophagy alone in microglia through deletion of FIP200 had little to no effect on the establishment of AD-like pathology when compared to autophagysufficient 5xFAD mice (Heckmann et al.,2019). Additionally,and as illustrated inFigure 1,we identified that a primary contributing factor to the decreased processing of internalized β-amyloid by LANDO-deficient microglia was not due to a defect in degradation,as is often seen in LAP-deficiency (Martinez et al.,2015; Martinez et al.,2016),but was rather caused by an impaired recycling of receptors that recognize β-amyloid,including TLR4 and TREM2 (Heckmann et al.,2019). Moreover,inflammatory cytokine production in response to β-amyloid paralleled the status of receptor recycling (Figure 1) (Heckmann et al.,2019). The role of the autophagy machinery in inflammatory pathways will be further described below.
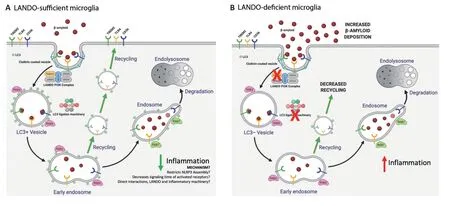
Figure 1|LANDO sufficiency or deficiency in microglia.
Impaired Noncanonical Functions of the Autophagy Machinery is Enough to Drive Alzheimer’s Disease
Since the AD phenotype associated with LANDO-deficiency was so greatly exacerbated,we decided to ask if LANDO-deficiency alone could drive AD-like pathology in mice in the absence of a humanized transgene(s). Elegant studies evaluating the WD-domain of the autophagy protein ATG16L and the binding regions required for interaction of ATG16L with the autophagy regulator WIPI2,led to the novel identification of a deletion mutant of ATG16L WD-domain that was sufficient for canonical autophagy but had an impairment of non-canonical pathways including LAP (Fletcher et al.,2018; Rai et al.,2019). We found that primary microglia lacking the WD-domain of ATG16L were likewise deficient in LANDO and presented with a severe impairment in the recycling of TREM2,TLR4,and CD36 (Heckmann et al.,2020b). Furthermore,those microglia had a decreased capacity for the continued uptake of extracellular β-amyloid (Heckmann et al.,2020b).
Evaluation of ATG16L WD-domain deficient mice that had been aged to 2 years revealed a deposition of endogenous murine β-amyloid in the hippocampus and cortex,consisting of neurotoxic β-amyloid 1-40 and 1-42 species (Heckmann et al.,2020b). When compared to β-amyloid deposits from LANDO-deficient mice on the 5xFAD background or human plaques,the deposits observed in the ATG16L WD-domain deficient mice were non-aggregated and more diffuse in morphology,consistent with the inherent biochemical and biophysical differences between mouse and human β-amyloid as reported previously (Lv et al.,2013). Nevertheless,these deposits of β-amyloid were neurotoxic and sufficient to drive downstream pathology including; tau hyperphosphorylation,reactive microgliosis and neuroinflammation,as well as active neurodegeneration (Heckmann et al.,2020b). Consistent with upstream markers of pathology,mice presented with severe memory and behavioral impairment.
Regulation of Inflammation by the Autophagy Machinery
While microglial LANDO suppresses neuroinflammation in response to β-amyloid,the role for other non-canonical uses of the autophagy machinery in the CNS in pathways such as LAP is less well defined. Currently,it is difficult to delineate LAP from LANDO at the genetic level (Martinez et al.,2015; Heckmann et al.,2017,2019; Heckmann and Green,2019). In pathologies characterized by neurodegeneration such as AD,LAP likely functions to prevent inflammation in response to the removal of dying neurons,a process called efferocytosis (Boada-Romero et al.,2020; Doran et al.,2020). Ongoing studies are directed at the genetic delineation of LAP,LANDO,and other non-canonical pathways. Having the ability to differentiate LAP and LANDO will allow for future studies that are directed at the contribution of each pathway to immune function and microglial responses to perturbations such as β-amyloid.
Peripheral to the CNS,non-canonical functions of the autophagy machinery have been shown to regulate inflammatory polarization of macrophages in response to a variety of cargoes (Heckmann et al.,2017; Heckmann and Green,2019). A substantial amount of effort has been directed at understanding how the autophagy machinery in LAP suppresses inflammatory activation in response to dead or dying cells (Green et al.,2016; Asare et al.,2020; Boada-Romero et al.,2020). Signals that promote the identification of dead cells,often referred to as “Find-me” signals as well as signals promoting the engulfment of dead cells,“Eat-me” signals,have been shown to help shape the macrophage response to dead cell cargo as reviewed previously (Heckmann et al.,2017; Heckmann and Green,2019; Boada-Romero et al.,2020). In addition,components of the autophagy machinery that are required for pathways like LAP and LANDO,such as ATG16L,have been shown to directly regulate antiinflammatory signaling (Martinez et al.,2015; Heckmann et al.,2019,2020b). The WD-domain of ATG16L interacts directly with cytokine receptors including IL-10RB and IL-2Rγ and exhibits WD-domain dependent LC3 lipidation (Serramito-Gomez et al.,2020). Cells deficient in the WD-domain of ATG16L have decreased anti-inflammatory signaling due to delayed endocytosis and insufficient trafficking of IL-10/IL-10R complexes leading to pro-inflammatory activation (Sorbara et al.,2013; Serramito-Gomez et al.,2020; Wang et al.,2021). Consistent with data on ATG16L,loss of Rubicon likewise alters IL-10 production and signaling (Martinez et al.,2016; Cunha et al.,2018). Whether Rubicon functions through downstream ATG16L or independently of the downstream components of LAP in regulating IL-10 biology remains unknown,although phenotypically,loss of either protein results in consistent changes to inflammatory polarization of macrophages. Moreover,stimuli that initiate these processes appear to be quite diverse,ranging from amyloids and extracellular aggregates to bacteria,viruses,and other pathogens (Heckmann et al.,2017; Heckmann and Green,2019). In all scenarios the non-canonical use of the autophagy machinery is functioning to suppress inflammation. Once loss of components required for LAP and LANDO occurs,it seems cells subsequently are primed towards the production of inflammatory cytokines.
Exploiting Neuroinflammation as a Therapeutic Avenue in Alzheimer’s Disease
One aspect we observed in the brains of the ATG16L WD-domain deficient aged mice was high levels of the inflammatory cytokine IL-1β in addition to TNFα,again paralleling human disease and consistent with inflammatory signaling described above. Over the past few years,there has been an accumulating interest in the possibility of targeting neuroinflammation as a therapeutic avenue for a variety of neurodegenerative conditions including AD. Targeting inflammatory cytokine production and/or signaling for mediators including IL-1β,TNFɑ,and IL-6 have been proposed (Camargo et al.,2015; Wu et al.,2015; Bronzuoli et al.,2016; Kinney et al.,2018; Lonnemann et al.,2020). In particular,IL-1β has proven to be an intriguing,albeit challenging target for putative therapy. Previous studies have demonstrated that compounds which modulate the activity of the NLRP3 inflammasome,the complex required for the production of IL-1β (Coll et al.,2019; Swanson et al.,2019; Jiang et al.,2020; Jiao et al.,2020),are efficacious and can readily cross the blood-brain barrier. Using our novel age-associated spontaneous AD model,we treated ATG16L WD-domain deficient mice with established AD-like disease for 8-weeks with the NLRP3 inhibitor MCC950 to reduce IL-1β production.Following therapeutic intervention,MCC950 treated mice had comparable levels of β-amyloid to those observed in vehicle treated animals (Heckmann et al.,2020b). However,there were robust decreases in microglia activation,neuroinflammation,tau phosphorylation,and prominent decreases in activate neuronal apoptosis (Heckmann et al.,2020b). Although optimistic,we were surprised to find that MCC950 treatment led to an approximate 80-90% restoration in behavior and memory capacity as evaluated by Y-maze and novel object recognition analyses compared to mice treated with vehicle,which continued to have a decline in memory from the onset of therapy (Heckmann et al.,2020b). Together,these findings suggest that neuroinflammation is upstream of neurodegeneration and is leading to neuronal death by “fire” or as a consequence of inflammatory activation. Our results suggest that by alleviating inflammatory processes in the AD brain,there is the potential to overcome neuronal loss and have a positive impact on memory and reduction in disease progression. Moreover,these findings confirm that targeting neuroinflammatory mediators such as IL-1β in an established disease model is a promising therapeutic approach.
The Path Ahead
Superficially our findings reiterate the importance of neuroinflammation to disease processes in AD and that neuroinflammation is central to disease establishment and progression. As such,neuroinflammation is an attractive therapeutic target not only in AD but in a plethora of other CNS pathologies including Huntington’s disease,Parkinson’s disease (PD),and amyotrophic lateral sclerosis (Rizzo et al.,2014; Liu and Wang,2017; Kwon and Koh,2020). In particular,neuroinflammatory modulation has been proposed in amyotrophic lateral sclerosis and PD with promising efficacy (Lu and Hu,2012; Guan and Han,2020),The role of LANDO or other non-canonical pathways using the autophagy machinery in these diseases has to date not been evaluated,although changes in receptor recycling in diseases such as PD hint at a plausible role for LANDO (Kim et al.,2013; Ferguson and Green,2014; Martinez et al.,2016; Muniz-Feliciano et al.,2017). Our results have further led to more in-depth biological questions,including how is LANDO-deficiency altering the neuroimmune architecture and by what mechanism(s)? Additionally,as hinted earlier,targeting IL-1β through inflammasome inhibition has proven more challenging in humans,with many compounds including MCC950 having deleterious side effects including hepatotoxicity. Similarly,targeting other inflammatory mediators such as TNFɑ which is also elevated in models of AD has proven less efficacious in human patients (Chang et al.,2017; Ekert et al.,2018). Would targeting non-canonical functions of the autophagy machinery in pathways such as LANDO prove less detrimental to peripheral systems while not sacrificing therapeutic efficacy? We can take hints from the role of pathways such as LAP in regulating inflammation in peripheral macrophages where the function appears to be fully independent of canonical autophagy (Martinez et al.,2015,2016; Heckmann et al.,2017; Heckmann and Green,2019),and as described above. A complete delineation from canonical autophagy would be ideal,as it allows for more targeted approaches to be applied to pathways such as LAP and LANDO. Previously,we have shown that abrogation of microglial autophagy in the 5xFAD model appears to have little to no effect on disease pathology and outcome (Heckmann et al.,2019). These data suggest that there is distinct regulation of LAP and LANDO compared to canonical autophagy and that each pathway likely is functioning independent of the others,although further investigation will be necessary to fully characterize these hypotheses.
Along those lines,we as well as others have shown that there is a consistent downregulation of components of the autophagy machinery and LANDO with age and further decreases in expression observed in the AD brain. Moreover,the suppression of pathways such as LANDO may prime the brain towards chronic neuroinflammatory activation,putatively “seeding” the brain for disease as one ages. With advances in genetic therapies and CRISPR technology,it is no longer unimaginable that we will soon have the capability of targeting these upstream regulators and associated pathways in human patients.
In summary,our prior work illustrates an important role for the autophagy machinery in non-canonical functions including LC3-associated endocytosis. Moreover,it is clear that LANDO is important for protection against neuroinflammation and downstream pathology in response to β-amyloid. Further studies are certainly required to unravel the exciting questions that are raised following the discovery of LANDO and its initial characterization in mitigating neuroinflammation and neurodegeneration,at both the cellular and molecular levels.
Author contributions:ADR and AH co-drafted the manuscript and were equally responsible for the collection and review of the literature cited herein. BLH revised and edited the manuscript and was responsible for final approval. All authors approved the final version of the manuscript.
Conflicts of interest:BLH is a co-inventor on United States patent applications; 62/795,217,filed January 22,2019,62/797,564 filed January 28,2019,and worldwide patent application PCT/IB2020/050504 filed January 22,2020,through the United States Patent and Trademark Office. BLH consults for Ventus Therapeutics and USA Prime Biotech.
Financial support:This work was supported in part by the funding from the National Institute of Allergy and Infectious Disease and the National Cancer Institute under award numbers AI138492 and CA231423 to BLH. The content herein is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Shouheng Jin,Sun Yat-sen University,China.
Additional file:Open peer review report 1.
Retraction
Retraction: Valproic acid protects neurons and promotes neuronal regeneration after brachial plexus avulsion
https://doi.org/10.4103/1673-5374.317993
Concerns have been raised about the western blot bands presented in Figures 2A and 4A of this article (Li et al.,2013; doi: 10.3969/j.issn.1673-5374.2013.30.006).
· Figure 2A appears to include splicing between lanes 1 and 2 in Bcl-2 blots in the valproic acid group.
· Figure 4A appears to include splicing between lanes 1 and 2,4 and 5,5 and 6 in c-Jun blots in the injury group.
The corresponding author has stated there was an error in the selection of blot images used for Figures 2A and 4A.
TheNeural Regeneration ResearchEditors have been unable to verify the accuracy and reliability of the western blot data presented in the paper. In light of the extent of the unavailability of the underlying original western blot data and the unresolved concerns regarding the accuracy and reliability of the figures,theNeural Regeneration ResearchEditors retract this article.
All authors agreed with the retraction.
Reference
Li Q,Wu D,Li R,Zhu X,Cui S (2013) Valproic acid protects neurons and promotes neuronal regeneration after brachial plexus avulsion. Neural Regen Res 8(30):2838-2348.
杂志排行

中国神经再生研究(英文版)的其它文章
- A Drosophila perspective on retina functions and dysfunctions
- Celeboxib-mediated neuroprotection in focal cerebral ischemia: an interplay between unfolded protein response and inflammation
- Pramipexole,a dopamine D3/D2 receptor-preferring agonist,attenuates reserpine-induced fibromyalgia-like model in mice
- Effects of delayed repair of peripheral nerve injury on the spatial distribution of motor endplates in target muscle
- Neurorehabilitation using a voluntary driven exoskeletal robot improves trunk function in patients with chronic spinal cord injury: a single-arm study
- Gene and protein expression profiles of olfactory ensheathing cells from olfactory bulb versus olfactory mucosa