Comprehensive silk gland multi-omics comparison illuminates two alternative mechanisms in silkworm heterosis
2022-08-05HanXuLeiChenXiaoLingTongHaiHuLiYuanLiuGuiChunLiuYaNanZhuRuoPingZhaoWenWang3FangYinDaiXinLiHuiXiang2
Han Xu ,Lei Chen ,Xiao-Ling Tong ,Hai Hu ,Li-Yuan Liu ,Gui-Chun Liu ,Ya-Nan Zhu,Ruo-Ping Zhao,Wen Wang3,,Fang-Yin Dai,*,Xin Li,Hui Xiang2,,*
1 School of Medicine, Shenzhen Campus of Sun Yat-Sen University, Shenzhen, Guangdong 518107, China
2 Guangdong Provincial Key Laboratory of Insect Developmental Biology and Applied Technology, Guangzhou Key Laboratory of Insect Development Regulation and Application Research, Institute of Insect Science and Technology, School of Life Sciences, South China Normal University, Guangzhou, Guangdong 510631, China
3 School of Ecology and Environment, Northwestern Polytechnical University, Xi’an, Shaanxi 710072, China
4 State Key Laboratory of Silkworm Genome Biology, Key Laboratory of Sericultural Biology and Genetic Breeding, Ministry of Agriculture and Rural Affairs, College of Biotechnology, Southwest University, Chongqing 400715, China
5 State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming,Yunnan 650223, China
ABSTRACT Heterosis is a common phenomenon in plants and animals with diverse underlying mechanisms.Here,we applied two widely used silkworm hybrid systems and performed multi-omics analysis to identify possible intrinsic associations between different hybrid strategies and epigenetic mechanisms with silkworm heterosis.We found significant differences in the silk gland transcriptomic landscape between the two systems,including differentially expressed genes and expression patterns in the hybrid offspring compared to their parents.In the quaternary hybrid system,hybrid vigor was primarily due to upregulated genes and the parent-dominant upregulated expression pattern,involving multiple transport processes,cellular nitrogen compound catabolism,glucose metabolism,and tricarboxylic acid cycle.In the binary system,hybrid vigor was mainly due to the down-regulated genes and transgressively down-regulated expression pattern,mainly involving basic nitrogen synthesis metabolism and body function.We also demonstrated that DNA methylation may affect hybrid vigor by regulating the expression of several heterosis-related genes.Thus,this study revealed two alternative mechanisms that may contribute to silkworm heterosis,both of which facilitate the efficient utilization of energy and nitrogen for silk production.
Keywords:Heterosis;Silkworm;Hybrid systems;Multi-omics;Silk production
INTRODUCTION
Heterosis,also known as hybrid vigor,is a well-known phenomenon whereby F1 hybrid offspring exhibit greater vigor than their inbred parents.Heterosis has long been used in agricultural production and domestic breeding (Dyer,1877;Fu et al.,2014;Goddard,2012;Shull,1948),and is an important genetic process in modern agriculture,allowing the production of new plant and animal varieties with high yield and quality.However,in contrast to the widespread application of heterosis,the genetic basis underpinning this phenomenon remains poorly understood.
Classical quantitative genetics attempted to explain heterosis based on the dominance hypothesis (Davenport,1908),overdominance hypothesis (East,1936),and epistasis hypothesis (Li et al.,2001;Yu et al.,1997).Although these hypotheses are not mutually exclusive,it is difficult to generalize the complex phenomenon of heterosis (Birchler et al.,2003,2006).Advances in molecular biology and genomics have provided the opportunity to explore important molecular mechanisms.Various -omics approaches,such as transcriptomics (Klosinska et al.,2016;Kong et al.,2020;Liu et al.,2018;Luo et al.,2021;Springer &Stupar,2007;Wang et al.,2019),proteomics (Hu et al.,2017;Rockenbach et al.,2018),and epigenomics of parents and hybrids (Lauss et al.,2018;Li et al.,2018;Zhou et al.,2021),have made considerable efforts to understand,verify,and update the classical genetic hypotheses.Despite these advances at the molecular level,our understanding of heterosis remains unclear given that its complexity,strength,and underlying mechanisms can also vary among species,varieties,and traits (Wu et al.,2021).Furthermore,few studies have attempted to explore the possible intrinsic associations between different hybrid strategies and hybrid vigor.
The silkworm (Bombyx mori) is an economically important domesticated insect and an ideal model for exploring the above questions.The discovery and application of hybrid vigor in silkworm breeding can be traced back to the mid-twentieth century.Hybrid silkworms have substantial economic advantages in silk production,silk quality,and disease resistance (Qin et al.,2012).Different hybridization methods,e.g.,binary hybrids (i.e.,one crossing) and quaternary hybrids(i.e.,double crossing) (Singh et al.,2002),can produce different progeny vigor (Ge et al.,2020;Samami et al.,2019;Sharma &Bali,2019;Wang et al.,2015).Quaternary hybridization can induce a broader genetic base than binary hybridization,potentially with dominant and superior gene complementation and more interaction types.However,not all quaternary hybrid systems produce better vigor than binary systems,and they may also be more time-consuming and costly.
To date,considerable effort has been devoted to exploring the molecular mechanisms underpinning silkworm heterosis.In addition to identifying potential heterosis-related genes (Li et al.,2015;Wang et al.,2015;Xiao et al.,2020;Zhang et al.,2018),it has been suggested that protein processing,metabolic slowdown,energy accumulation,and silk protein expansion may account for silk yield heterosis (Zhang et al.,2018).Xiao et al.(2020) also suggested that although phenotypic indicators (e.g.,cocoon shell weight) of hybrid vigor may be overly dominant,the expression of many genes and proteins does not exceed and may be lower than that of the parents.Despite these studies,our understanding of the possible intrinsic associations between different hybrid strategies and silkworm heterosis remains limited.Furthermore,as an alternative mechanism in phenotypic evolution,epigenetics may also play a role in heterosis (Chen,2013).DNA methylation is a highly prevalent epigenetic marker in the genome and an important cause of heterosis(Lauss et al.,2018;Zhou et al.,2021).We previously demonstrated that the epigenetic system is functional in silkworms and played an important role in silkworm domestication (Xiang et al.,2010,2013).We further suspect that epigenetics may also be involved in silkworm hybridization and hybrid vigor.However,no studies have attempted to decipher silkworm heterosis from an epigenetic perspective.
In the present study,we used the silk gland as a target organ and explored the possible intrinsic associations between different hybrid strategies and epigenetic mechanisms with silkworm heterosis.
MATERIALS AND METHODS
Sample preparation and phenotypic trait measurement
In this study,we designed two different Chinese-Japanese hybrid systems,i.e.,quaternary and binary hybridization.For the quaternary hybrid system,we selected silkworms from two Chinese strains (CF0) and two Japanese strains (JF0) for inbreeding,respectively,and then crossed the F1 female individuals of the Japanese inbred progeny (JF1) with the F1 male individuals of the Chinese inbred progeny (CF1) to produce quaternary hybrid silkworms (F2,Figure 1A).Each group (CF0,JF0,JF1,CF1,and F2) contained not less than 30 female and 30 male silkworms,which were raised under controlled temperature and humidity conditions with the same mulberry leaf intake.On the fourth day of the fifth instar silkworms,total cocoons and cocoon shells were weighed.Silk glands were collected and stored at −80 °C (one female and one male from each of the five groups) for downstream experiments.
For the binary hybrid system,we crossed a female Chinese strain silkworm (CF0) with a male Japanese strain silkworm(JF0) to breed binary hybrid silkworms (F1,Figure 1A).The two strains used were two of the strains used in the quaternary system.On the fourth day of the fifth instar silkworms,total cocoons and cocoon shells were weighed.Silk glands of four individuals (CF0,JF0,one female,and one male from F1 group) were collected and stored at −80 °C for downstream experiments.
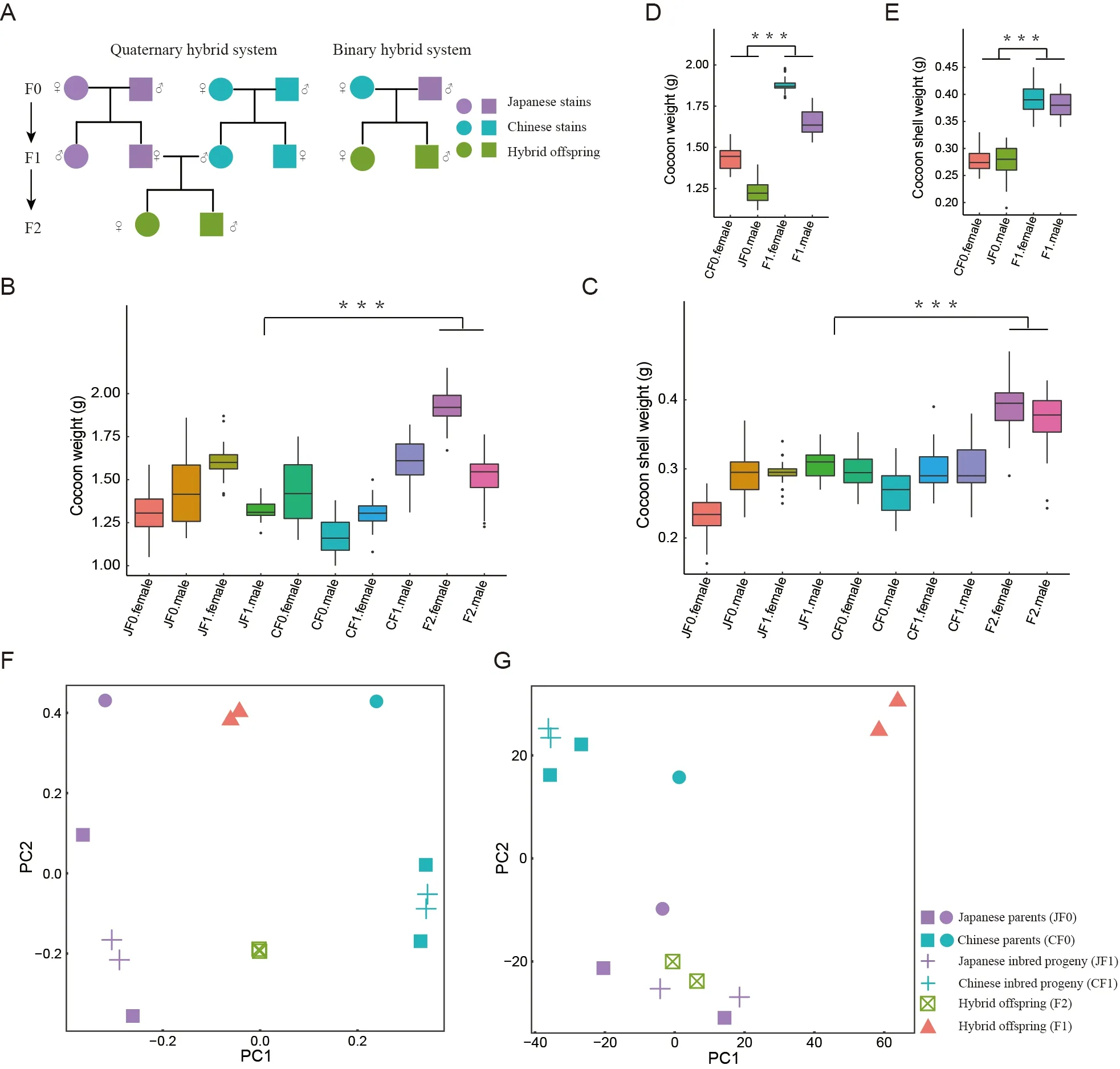
Figure 1 Offspring of two hybrid systems showed significant hybrid vigor
DNA and RNA extraction,library construction,and sequencing
The silk glands were ground into powder in liquid nitrogen.Half of the powder from each silk gland was used for total DNA extraction with a DNeasy Blood &Tissue Kit (QIAGEN,Germany),while the remaining powder was used for RNA extraction with a RNeasy Mini Kit (QIAGEN,Germany)according to the manufacturers’ instructions.Whole-genome sequencing (WGS),RNA-sequencing (RNA-seq),and wholegenome bisulfite sequencing (WGBS) libraries were constructed and sequenced by Novogene (China).
WGS data processing
Raw reads were subjected to quality control using Trimmomatic (v0.39) (Bolger et al.,2014) and were aligned to the silkworm reference genome using Burrows-Wheeler Aligner (BWA) software (v0.7.12) (Li &Durbin,2009).The silkworm (B.mori) reference genome and genome annotation information were obtained from the DEYAD platform(https://doi.org/10.5061/dryad.fn82qp6).The Genome Analysis Toolkit (GATK;v3.8) was applied for indel realignment and variant calling following the recommended best practice protocols by HaplotypeCaller.High-confidence single nucleotide polymorphisms (SNPs) were obtained by filtering raw vcf files using the parameters “QUAL<30 || DP<3|| QD<2 || FS>=30” (Depristo et al.,2011).To improve the mapping rate,a pseudogenome was constructed by replacing both homozygous and heterozygous loci with alternative SNPs in the reference genome.
RNA-seq data processing
Raw reads were subjected to quality control using Trimmomatic (v0.39),and were aligned to the silkworm reference genome using Hisat2 (v0.1.5-beta) (Kim et al.,2015).Gene expression levels were quantified using Stringtie v2.0.4 (Pertea et al.,2015) and normalized using the fragments per kilobase of transcript per million fragments mapped (FPKM) method.The DESeq and DESeq2 R language (v3.6.1) packages were used to calculate differentially expressed genes (DEGs) for groups without and with biological duplication,respectively.The DEGs were selected based onP<0.05 and |log2FC (fold-change)|>1 criteria.Binary hybrid system parents (CF0 and JF0) were processed using GATK (v3.3) to generate raw SNP files of the parents,following the RNA best practice protocols.The raw SNP files were then filtered to generate parent genotypes with the parameters “QUAL<30 || DP<10 || QD<2 || FS>=30”.
Inherited expression mode identification
Gene expression modes of inheritance were assigned by comparing expression levels between maternal parent,paternal parent,and their offspring.Genes exhibiting <2-fold change in expression were designated as conserved genes(F1=M=P,where F1,M,and P represent expression levels of offspring,maternal parent,and paternal parent,respectively).Genes showing >2-fold change in expression between hybrids and either parent were regarded as differentially expressed pattern genes (DEPGs) (McManus et al.,2010).If the expression level of a DEPG in offspring was higher than one parent but lower than the other,the DEPG was defined as additive;if the expression level of a DEPG in offspring was similar to either parent,the DEPG was defined as dominant;if the expression level of a DEPG in offspring was higher or lower than the parents at the same time,the DEPG was defined as transgressive.Based on the above criteria,eight types of DEPGs were identified based on expression patterns compared to parents,i.e.,TS (Transgressive Up,F1>M and F1>P),TD (Transgressive Down,F1
Analysis of allele-specific expression (ASE) genes
Genotypes of the hybrid individuals and their parents were obtained after data processing.We selected heterozygous loci to determine ASE as they could be distinguished from paternal and maternal alleles.To obtain reliable results,we excluded heterozygous loci with total mapped reads <20.For each reliable heterozygous site,we used RNA-seq mapping files to count reads supporting the paternal or maternal alleles.The parental allelic ratio was calculated as the number of parental allelic reads divided by the total number of mapped reads at a heterozygous locus.This ratio was adjusted to zero if the number of mapped reads was less than 2.The ASE sites were identified if the parental allelic ratio was <0.3 or >0.7.We defined genes with three or more ASE sites as ASE genes.All data processing was conducted using in-house Perl scripts.
WGBS data processing
For WGBS data processing,adapter and low-quality reads were first trimmed.The clean reads were then mapped to the pseudogenome using Bismark (v0.22.1),allowing up to two mismatches (Krueger &Andrews,2011).Unique mapped reads were extracted to determine the cytosine methylation level,as described previously (Lister et al.,2008),and only those cytosines with more than four reads in a library were used.The conversion rate was calculated based on the methylation level of non-CG sites,and the binomial test was used to correct the CG methylation level following previous research (Lister et al.,2009).The DSS package (v2.38.0) in R was used to calculate differentially methylated regions (DMRs)and bedtools (v2.29.0) was used to annotate the DMRs.
Functional annotation of candidate genes
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of candidate genes were performed using OmicShare tools,an online platform for data analysis (https://www.omicshare.com/tools).
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) validation of binary system genes
We performed qRT-PCR for certain binary system genes.Primers were designed using Oligo software (v7.60).Three biological replicates and three experimental duplicates were carried out for parents and hybrids (male,female).The relative expression values (2-ΔCt) of selected genes were used to compare parents and F1 hybrids.
RESULTS
Hybrids in quaternary and binary systems both showed obvious heterosis
We used two hybrid systems widely applied in traditional silk and cocoon production (Figure 1A).In the quaternary hybrid system,total cocoon weight and cocoon shell weight of the F2 hybrids were significantly higher than those of the pure F0 and inbred F1 varieties (P<0.001,two-tailedt-test,Figure 1B,C).However,there was no significant difference in cocoon shell weight between the inbred F1 generation and F0 generation,suggesting that a certain genetic distance between parents is necessary for hybrid vigor.In the binary hybrid system,in which a female Chinese strain (CF0) and male Japanese strain (JF0) were used to generate hybrid offspring,total cocoon weight and cocoon shell weight of the F1 hybrids were significantly higher than those of their parents (P<0.001,twotailedt-test,Figure 1D,E).Thus,we constructed two different hybrid systems,both of which showed obvious heterosis.
Principal component analysis (PCA) showed different gene expression landscapes between hybrid systems
To investigate the mechanisms underlying hybrid vigor,we performed WGS and RNA-seq for individuals in both hybrid systems.After quality control,39 579 515–53 344 752 and 45 094 910–93 497 243 clean reads were obtained from WGS and RNA-seq,respectively.The mapping rates for the WGS and RNA-seq reads were 81.24%–88.87% (Supplementary Table S1) and 57.90%–84.77% (Supplementary Table S2),respectively.SNP-based PCA showed that the Japanese and Chinese strains were clearly separated in both hybrid systems.As expected,the hybrid individuals were located between their parents in both hybrid systems (Figure 1F).Consistently,gene expression-based PCA revealed a clear separation between the Japanese and Chinese strains.However,the hybrid progeny exhibited very different patterns at the transcriptome level compared to the SNP-based patterns.In the binary system,hybrid individuals were located far from both parents,while quaternary system hybrids were clustered together with their maternal Japanese strain(Figure 1G).These results suggest significantly distinct gene expression patterns between the systems,implying that different molecular mechanisms may be involved in the two hybrid systems.
Hybrid vigor was associated with many functional DEGs between parents and hybrid offspring
DEGs play an important role in the formation of hybrid vigor(Ge et al.,2020;Wang et al.,2015).Here,we identified DEGs in both the quaternary and binary systems.The RNA-seq results were successfully validated by real-time PCR of several genes (Supplementary Figure S1).
In the quaternary system,few DEGs were found between the inbred offspring and their parents,regardless of the Chinese (90 DEGs) or Japanese (42 DEGs) strains.In contrast,many DEGs (679) were found in the hybrid F2 offspring,including 374 up-regulated and 305 down-regulated DEGs (Figure 2A,B;Supplementary Table S3).These results suggest that the F2 hybrids showed significant heterosis coupled with marked transcriptome changes compared to their parents.In addition,the DEGs between the F2 hybrids and both parents largely overlapped with the DEGs between the F2 hybrids and their Chinese parent (CF1) (Supplementary Figure S2A,B),implying that they were more divergent from the Chinese side,consistent with the transcriptomic PCA results showing that quaternary system hybrids were clustered with Japanese strains.
To determine whether the DEGs between F2 hybrids and F1 parents are associated with divergence between the Japanese and Chinese strains,we compared DEGs between the strains in both F0 and F1 generations,respectively,and identified only 21 shared DEGs (Supplementary Figure S2C,D).These results suggest that the DEGs in hybrid individuals did not arise directly from divergence between parental strains but may have originated from potential epistasis in different strains.
In the binary system,we identified many DEGs (638 upregulated and 966 down-regulated) between the hybrid offspring and their parents,again suggesting that the large number of DEGs may have contributed to hybrid vigor(Figure 2C,D;Supplementary Table S3).Notably,significantly more genes were down-regulated than up-regulated,indicating that gene repression may be important for the occurrence of heterosis in the binary system.Interestingly,unlike the quaternary system results,DEGs in the F0_vs_F1 group largely overlapped with the CF0_vs_F1 and JF0_vs_F1 groups (Supplementary Figure S2E,F),suggesting that the underlying mechanisms of hybrid vigor differ between the two systems.We also compared DEGs between the different stains (CF0_vs_JF0) and the F0_vs_F1 group and detected only four up-regulated and three down-regulated overlapping DEGs (Supplementary Figure S2G,H),thus highlighting the important role of epistasis in hybrid vigor formation,as reported previously (Li et al.,2001;Yu et al.,1997).
In both hybrid systems,only 25 up-regulated and 44 downregulated DEGs were shared between hybrid offspring and their parents (Figure 2E,F).Therefore,although both hybrid systems showed significant hybrid vigor,the gene sets involved in heterosis formation may be quite different between the two systems.We performed GO and KEGG enrichment analyses of DEGs between hybrid individuals and their parents.Results showed that the up-regulated DEGs in the quaternary system were mainly involved in “carbon metabolism”,“citrate cycle (TCA cycle)”,and biological processes related to protein transportation (Figure 2G),whereas the down-regulated DEGs were involved in many processes related to molecular binding (Figure 2H;Supplementary Tables S4,S5).These findings suggest that those up-regulated DEGs related to protein transport and energy supply significantly contributed to heterosis in the quaternary system.In contrast,enrichment analysis of the binary system showed that the up-regulated DEGs were mainly involved in “retinol metabolism” and “choline metabolism and longevity regulating pathway” (Figure 2G),while the down-regulated DEGs were mainly involved in “gene expression”,“translation”,and “ribosome biogenesis and DNA replication” (Figure 2H;Supplementary Tables S4,S5).These results indicate that binary system heterosis is generally a lower metabolic rate process,which may help concentrate energy for silk production.Taken together,the two systems showed distinct DEGs and functional enrichment,suggesting different underlying mechanisms of hybrid vigor.
Different hybrid systems exhibited unique gene expression patterns
DEPGs play an important role in the formation of hybrid vigor in many species and exhibit strong species and tissue specificity (McManus et al.,2010;Xiao et al.,2020).In the quaternary system,we identified 543 DEPGs in the Japanese F1 inbred individuals and 248 DEPGs in the Chinese F1 inbred individuals,with PDD being the most abundant pattern(Supplementary Figure S3A,B).In the hybrid F2 offspring,we detected 1 699 DEPGs in the hybrid individuals,with PDU being the most abundant pattern (652 genes,38.38%)(Figure 3A;Supplementary Tables S3,S6),suggesting that these DEPGs,especially PDU genes,play an important role in the formation of hybrid vigor in the quaternary system.GO and KEGG analysis of the PDU genes indicated enrichment in“intracellular protein transport”,“protein transport”,“intracellular transport” and “protein processing in endoplasmic reticulum” (Figure 3B;Supplementary Tables S4,S5),similar to the function of the above up-regulated DEGs between hybrids and their parents,thus suggesting that PDU genes related to protein transport and processing are important for quaternary system heterosis.We also compared the upregulated DEGs and PDU gene sets,but only found 34 overlapping genes (Supplementary Figure S3C).Although the two gene sets shared very few genes,they showed similar biological functions,emphasizing the importance of protein synthesis and transport in quaternary system hybrid vigor.

Figure 2 Comparisons of DEGs between hybrid offspring and their parents in two hybrid systems
In the binary system,we identified 1 613 DEPGs,with TD being the most abundant pattern (803 genes,49.78%)(Figure 3C;Supplementary Table S6).The TD genes were enriched in “gene expression”,“translation”,and “ribosome biogenesis and DNA replication”,consistent with the downregulated DEGs found in the binary hybrids (Figure 3D;Supplementary Tables S4,S5).In addition,a relatively large proportion of TD genes (655/803,81.57%) were shared with the down-regulated DEGs (655/966,67.81%) (Supplementary Figure S3D),suggesting that repression of certain genes(down-regulated DEGs and TD genes) may be important for binary system hybrid vigor and that hybrid vigor may be associated with lower metabolic rates but possibly higher metabolic efficiency in the binary system.
Results showed that MDD was the second most abundant pattern in both hybrid systems,accounting for 22.31% (379)and 22.01% (355) of genes,respectively.However,only 22 genes overlapped between them (Figure 3E) and the two MDD gene sets were enriched in distinct biological processes(Figure 3F).Thus,based on DEPG analysis,the molecular mechanisms of hybrid vigor appear to differ significantly and may recruit very different gene sets and biological pathways to facilitate similar heterosis phenotypes.
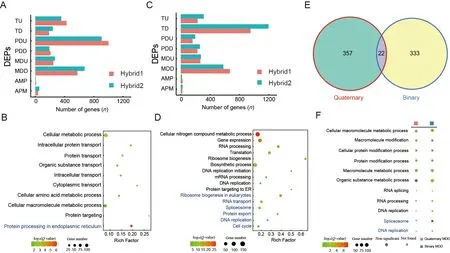
Figure 3 Differences in DEPGs between two hybrid systems
ASE may be dispensable for silkworm hybrid vigor
ASE genes are known to contribute to the formation of hybrid vigor in rice and maize (Guo et al.,2008;Shao et al.,2019;Springer &Stupar,2007).To investigate whether ASE genes impact hybridization superiority in silkworm,we determined ASE gene number in the quaternary system hybrid individuals.We obtained 619 ASE genes but only a few showed biased expression patterns,including 67 PDU genes (16.50%) and 29 MDD genes (7.14%) (Supplementary Figure S4A and Table S7).We also found no significant enrichment of ASE genes in the different DEPG groups (P=0.243,chi-square test).In addition,no obvious bias was observed between DEPGs and maternal or paternal ASE genes (P=0.243,P=0.230,chisquare test;Supplementary Figure S4B),indicating that ASE genes were evenly distributed across the DEPG groups.These results further suggest that ASE genes have little effect on gene expression patterns and are dispensable in the formation of hybrid vigor.
Genes showing changes in DNA methylation in hybrid offspring were related to silk gland metabolism and protein synthesis
Methylated cytosines in theB.morigenome are mainly enriched in the body region of the genes and have a significant regulatory effect on genes in the silk gland tissue(Xiang et al.,2010).To explore the contribution of DNA methylation to hybrid vigor,we investigated the DNA methylomes of parents and F1 and F2 offspring of the quaternary system,yielding 13.52–19.45 Gb methyl-seq data and 65.74%–73.92% of CpG sites with an average sequencing depth of 10 x per CpG cytosine (Supplementary Table S8).DNA methylation-based PCA of gene bodies in each quaternary hybrid individual showed a clear separation between Chinese and Japanese strains.The F2 hybrid vigor individuals were mostly located between their parents,albeit slightly closer to the Japanese parent,partially consistent with the SNP-based results.These results suggest that the global DNA methylation landscape is mainly determined by genomic information (Figure 4A–C).
To explore whether hybrid vigor is associated with changes in DNA methylation,we compared inbred individuals with their Japanese and Chinese strain parents (JF0_vs_JF1,CF0_vs_CF1) and obtained 135 and 262 genes with differentially methylated regions (DMRs) in the gene body region,respectively.We further compared hybrid individuals with their parents (F1_vs_F2,JF1_vs_F2,and CF1_vs_F2)and obtained 391,459,and 653 genes with DMRs,respectively,much higher than the number of genes that differed between inbred individuals and their parents(Figure 4D).Although annotation of the promoter regions of the silkworm genome was incomplete,we found a similar trend in the promoter regions as in the gene bodies(Figure 4E),suggesting that DNA methylation changes in many genes of the hybrid F2 generation may play an important role in the formation of heterosis.To determine whether the source of DMRs in the F1_vs_F2 group may be related to differences between strains,we compared strains in both the F0 and F1 generations.Results showed a considerable proportion of genes with DMRs in the gene body(accounting for 43.2% in the F1_vs_F2 group,169 genes)between F1_vs_F2 and the other two groups (Figure 4F),suggesting that methylation differences between the strains may be an important reason for differences in the hybrids compared to their parents.

Figure 4 Contribution of DNA methylation to hybrid vigor formation
To explore the genes with gene body DMRs between parents and hybrids,we merged genes in the F1_vs_F2,JF1_vs_F2,and CF1_vs_F2 groups for enrichment analysis.Results showed that these genes were involved in “gene expression”,“macromolecule biosynthetic process”,“translation”,“protein transport”,“ribosome”,“RNA transport”,and “spliceosome” (Supplementary Tables S4,S5),suggesting that these genes may contribute to hybrid vigor via changes in DNA methylation.
PDU genes GLEAN_00865 and GLEAN_05534 may be regulated by epigenetics and involved in protein transportation
As PDU genes played an important role in hybrid vigor in the quaternary system,we explored whether these genes are regulated by methylation levels.We overlapped the DMR genes in the JF1_vs_F2 group with the PDU genes,and identified 50 overlapping genes (Figure 5A),includingGLEAN_00865(coatomer protein complex subunit beta 2 isoform X2),which is responsible for vesicle transport between Golgi apparatus and endoplasmic reticulum (ER) (Feng et al.,2021),andGLEAN_05534(annulin isoform X2),which plays an important role in maintaining cell and intercellular superstructure stability as an invertebrate transglutaminase(Bastiani et al.,1992;Singer et al.,1992).The DMRs were located in the twelfth intron of theGLEAN_00865gene(Figure 5B) and the seventh exon of theGLEAN_05534gene(Figure 5C).The expression levels of the two genes and methylation levels of the DMRs were significantly reduced in the F2 hybrids compared to the female parents,but not significantly changed compared to the male parents(Figure 5D).GLEAN_00865transports cargo molecules,such as proteins and lipids,from the Golgi to the ER and mediates the reverse and forward transport of materials between the Golgi membrane and vesicles (Arakel &Schwappach,2018;Béthune &Wieland,2018);therefore,it plays an important role in the process of silk secretion in the silk gland.These results indicate thatGLEAN_00865andGLEAN_05534are typical PDU genes,which are regulated by epigenetics and play important roles in protein transport and silk secretion,suggesting that changes in the epigenome are also involved in hybrid vigor.
DISCUSSION
We used two different silkworm hybrid systems and confirmed that silk yield traits were over dominant in vigorous hybrids from both systems.Interestingly,however,we did not observe significantly higher expression of silk protein genes in the vigorous hybrid offspring (Supplementary Figure S5) in either system.This pattern suggests that silk yield heterosis may not be directly attributable to enhanced silk gene transcription but rather to post-transcriptional or translational processes.Another possibility may be that silk yield heterosis is not limited to the up-regulation of silk genes.Increased silk protein synthesis,as also proposed by Xiao et al.(2020),could be attributed to the rearrangement of genes involved in multiple catalysis and metabolism-related pathways.Therefore,different aspects may be intrinsically convergent in their contribution to silk yield heterosis.
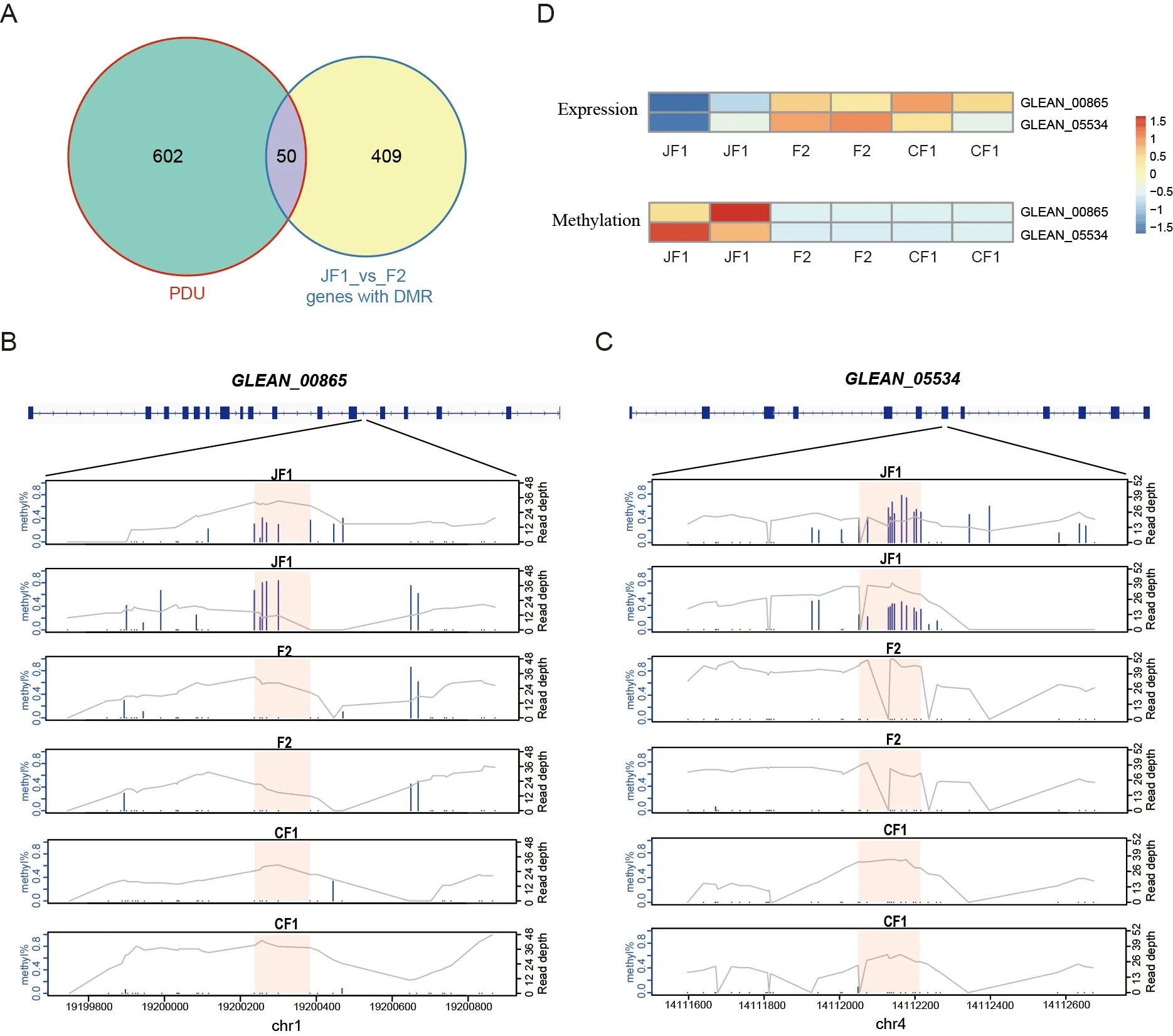
Figure 5 Two PDU genes involved in protein synthesis and transport are regulated by DNA methylation
Based on genomic,transcriptomic,and methylomic analyses of two different silkworm hybrid systems,we explored possible intrinsic associations between the different hybrid strategies and hybrid vigor.Results showed marked differences in the silk gland transcriptomic landscapes between the two systems,including differences in DEGs and DEPs between offspring and their parents.These findings reinforce the concept that the mechanisms underlying heterosis differ among different species and varieties (Zhou et al.,2021).As demonstrated above,enhanced gene expression in the quaternary system and overall repressed gene expression in the binary system alternatively participated in silk yield heterosis formation and convergently contributed to hybrid vigor.
In the quaternary system,increased gene expression,i.e.,up-regulated genes and PDU genes,may play a major role in the formation of silk yield heterosis.Although the two gene sets shared few genes,they were enriched in similar biological processes and pathways,such as protein transportation.Most proteins and peptides are transported to the ER or Golgi apparatus for further processing before performing their biological functions in cells (Krieg et al.,1989).Studies have shown that strains with high expression of genes related to the above pathways have higher silk yields (Wang et al.,2016).We also found that these genes were enriched in “cellular nitrogen compound catabolic process”,“glucose metabolic process”,and “tricarboxylic acid (TCA) cycle”.Glucose is central to energy consumption and carbohydrates,lipids,and proteins all ultimately break down into glucose.Glucose is the main precursor for the synthesis of different carbohydrates like glycogen,ribose,and deoxyribose,galactose,glycolipids,glycoproteins,and proteoglycans.At the cellular level,glucose is often the final substrate that enters a cell,where it is converted into adenosine triphosphate (ATP) to provide energy for many cellular processes.Within the mitochondrial matrix,the TCA cycle is a common metabolic pathway for all fuels and is responsible for the production of most reduced coenzymes used for the generation of ATP in the electron transfer chain (Akram,2014).Energy metabolism is necessary for silk production,and higher energy metabolism is associated with greater silk gland production (Wang et al.,2014,2016).Cellular nitrogen catabolism involves the breakdown of nitrogen into free ammonia,which can be further utilized in the silk gland for silk protein synthesis(Hirayama et al.,1997).Glucose metabolism and the TCA cycle provide energy and they also provide carbon skeletons for silk protein amino acids (Børsheim et al.,2004;Ni et al.,2015).Activation of efficient organic substance transportationrelated genes may enhance conveyance of nitrogen resources to the silk gland and conveyance of silk proteins outside the silk gland cells.In general,the above activation processes in vigorous hybrids may facilitate silk production and possibly lead to an increase in cocoon weight and cocoon shell weight.
However,the above processes were not activated in the binary system hybrids.Intriguingly,the hybrid offspring showed a nearly opposite landscape to the quaternary system in gene expression patterns,with significant repression of key processes that maintain nitrogen metabolism and body operation,such as peptide and amide biosynthesis,gene expression,translation,ribosomal biogenesis,RNA transportation,and DNA replication.This phenomenon has also been reported in a recent silkworm heterosis study (Xiao et al.,2020),indicating potential universality.As mentioned above,silk glands are highly differentiated,and proteins are highly specified in amino acid composition.Thus,we suspect that basic nitrogen metabolism for non-specific protein biosynthesis competes with silk protein production.Hence,these processes were strongly inhibited in the silk gland of the hybrid offspring to maximize protein production efficiency,resulting in an increase in cocoon weight and cocoon shell weight.However,how the repression of basic nitrogen metabolism triggers highly differentiated silk protein synthesis remains unclear.The biological processes enriched in the upregulated DEGs,such as “retinol metabolism”,and “choline metabolism and longevity regulating pathway”,may provide some cues.Retinol metabolism is crucial for many physiological processes,including embryonic development,reproduction,postnatal growth,differentiation,maintenance of epithelia,immune response,vision (Lidén &Eriksson,2006),glycogen storage (Saeed et al.,2020),and salivary gland initiation (Metzler et al.,2018).The salivary gland is homologous to the silk gland.Hence,the above enriched biological processes may play a role in silk production.Choline metabolism is also important in multiple processes and is involved in larval brain development in the tobacco hornwormMedusa sextan(Lester &Gilbert,1986).Silkworm brain is under strong artificial selection and play vital role in multiple processes (Cui et al.,2021).These results suggest that silk yield heterosis required systematic modifications in the silkworm rather than mere enhancement of silk glands.
Regarding the role of epigenetics in silk yield heterosis,we were unable to illustrate a clear contribution of DNA methylation to heterosis at the genomic scale.However,we were able to identify two PDU genes (i.e.,GLEAN_00865andGLEAN_05534) in the quaternary vigorous hybrid offspring,which carried gene body DMRs in comparison to their parents.TheGLEAN_00865gene carries proteins and lipids from the Golgi apparatus to the ER and mediates the reverse and forward transport of materials between the Golgi membrane and vesicles (Adolf et al.,2019;Béthune &Wieland,2018),which are important steps in silk and cocoon production.
Notably,key genes involved in the glutamate and aspartate acid metabolic pathways,which play important roles in the utilization of nitrogen for silk production (Xiang et al.,2018),showed conserved or up-regulated expression in the vigorous hybrids in both systems (Supplementary Figure S6).Therefore,we suspect that the efficiency of ultimately utilizing nitrogen for silk production is well maintained in both vigorous hybrids regardless of the upstream mechanism.
CONCLUSIONS
In this study,we illustrated two distinct silk gland transcriptome landscapes and mechanisms underlying heterosis in two silkworm hybrid systems.Enhancement of transport or inhibition of basic nitrogen metabolism and body functioning can alternatively result in silkworm heterosis,depending on the hybridization strategy.We also preliminarily ascertained the epigenetic landscape of silkworm heterosis and proposed two genes that may be under epigenetic regulation and involved in silkworm heterosis.Our results should advance research on heterosis and provide additional genetic resources and approaches to improve silk yield.
DATA AVAILABILITY
All sequences reported in this study were deposited in the Genome Sequence Archive database (http://gsa.big.ac.cn/)under accession No.CRA006878,in the Science Data Bank(https://www.scidb.cn/) under DOI:10.57760/sciencedb.j00139.00003,and in the NCBI database under BioProjectID PRJNA770282.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
H.X.(Hui Xiang) and X.L.designed and supervised the study;W.W.and F.Y.D.participated in the design of the experiment;H.X.(Han Xu) and L.C.carried out all analyses and wrote the manuscript;X.L.T.,H.H.,L.Y.L.,G.C.L.,Y.N.Z.,and R.P.Z.were responsible for silkworm breeding,index determination,and DNA and RNA extraction.All authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank all anonymous reviewers for their comments on the manuscript.
杂志排行

Zoological Research的其它文章
- Zoological Research call for papers of Cavefish Special Issue
- Fuel source shift or cost reduction:Context-dependent adaptation strategies in closely related Neodon fuscus and Lasiopodomys brandtii against hypoxia
- Ecological study of cave nectar bats reveals low risk of direct transmission of bat viruses to humans
- Population and conservation status of a transboundary group of black snub-nosed monkeys (Rhinopithecus strykeri) between China and Myanmar
- Nucleus accumbens-linked executive control networks mediating reversal learning in tree shrew brain
- Europe vs.China:Pholcus (Araneae,Pholcidae) from Yanshan-Taihang Mountains confirms uneven distribution of spiders in Eurasia