高效液相色谱法测定注射用左亚叶酸钠有关物质的研究
2022-08-01姜海兵
姜海兵,邱 文
(深圳市资福药业有限公司,广东 深圳 518120)
亚叶酸钠与5-氟尿嘧啶合用治疗胃癌和结直肠癌。亚叶酸是四氢叶酸(THF)的5-甲酰衍生物的非对映异构体混合物,其生物活性物质为左旋体称为左亚叶酸[1]。本文经试验研究采用HPLC法测定有关物质,重复性好,结果准确可靠[2-5]。
1 仪器和试药
1.1 仪 器
美国Thermo U3000高效液相色谱仪,包括U3000四元泵,U3000紫外检测器,U3000柱温箱和U3000自动进样器,变色龙工作站
1.2 试 药
试药:注射用左亚叶酸钠(规格:50 mg/瓶(以左亚叶酸计),批号121001、121002、121003,深圳市资福药业有限公司;亚叶酸钙对照品:中检所,批号:100252-200703,100252-200403;10-甲酰叶酸(EP标准品);甲醇、乙腈:色谱纯,天津四友,Honey well;其余试剂均为分析纯,广州化学试剂厂,天津大茂化学试剂厂。
2 试验方法和结果
2.1 溶液配制
取本品适量,加流动相制成每1 mL中约含1 mg的溶液滤过,弃去初滤液,取续滤液作为供试品溶液;
精密量取1 mL供试品溶液,置100 mL量瓶中,加流动相稀释至刻度,摇匀,作为对照溶液(1);
取10-甲酰叶酸对照品适量,置25 mL量瓶中,加流动相稀释至刻度,摇匀,作为对照品溶液储备液,精密量取1 mL对照品溶液储备液,置50 mL量瓶中,加流动相制成每1 mL含8 μg的溶液,作为对照液(2)。
2.2 液相色谱条件选择
2.2.1 流动相及色谱柱选择
流动相:10%四丁基氢氧化铵-甲醇-乙腈-水(37.5:15:190:750)用磷酸调节pH值至7.5±0.1。
色谱柱:以十八烷基硅烷键合硅胶为填充剂的液相色谱柱;本实验使用Kromasil,C18柱,5 μm,250 mm×4.6 mm。
2.2.2 检测波长的选择
按2.1制备供试液,按上述色谱条件进样,在190~900 nm进行全波长扫描,记录吸收图谱,结果表明在该色谱条件下,左亚叶酸及其有关物质在280 nm下有较好的吸收,因此优选280 nm作为有关物质测定波长。
2.3 专属性试验
2.3.1 已知杂质试验
精密称取左亚叶酸钠冻干粉及空白辅料适量(约相当于左亚叶酸50 mg),置于50 mL容量瓶中,精密量取10-甲酰叶酸对照品溶液储备液1 mL加入,加流动相稀释至刻度,摇匀,滤过,弃去初滤液,取续滤液20 μL,注入液相色谱仪,记录色谱图,结果表明左亚叶酸主峰与10-甲酰叶酸峰分离度大于2.5。
2.3.2 破坏试验
采用强光照射、高温、酸(碱)水解及氧化的方法对空白辅料及制剂进行加速破坏,用以考察可能存在的降解产物和降解途径。
分别精密称取左亚叶酸钠冻干粉及空白辅料适量(约相当于左亚叶酸50 mg),置于50 mL容量瓶中,加流动相溶解并稀释至刻度,摇匀,滤过,弃去初滤液。取续滤液作为供试品溶液,分别精密量取各续滤液20 μL,注入液相色谱仪,记录色谱图,以此作为破坏实验的阴性对照样品,考察各破坏实验的结果。
(1)光破坏试验
精密称取左亚叶酸钠冻干粉及空白辅料适量(约相当于左亚叶酸50 mg),置于50 mL容量瓶中,加入流动相适量,超声使溶解,在光照条件下(4500 lx)照射12 h以上,将各溶液稀释至刻度,制成浓度约为1 mg/mL(以左亚叶酸计)的样品溶液,滤过,弃去初滤液,精密量取续滤液20 μL,注入液相色谱仪,记录色谱图。结果表明:与阴性对照样品进行比较,光照对空白辅料及制剂均没有破坏作用。
(2)酸破坏试验
精密称取左亚叶酸钠冻干粉及空白辅料适量(约相当于左亚叶酸50 mg),置于50 mL容量瓶中,加入0.1 mol/L盐酸溶液至刻度,超声使溶解。水浴加热2 h,放冷,滤过,弃去初滤液。精密量取续滤液以流动相稀释制成浓度约为1 mg/mL(以左亚叶酸计)的样品溶液,精密量取20 μL,注入液相色谱仪,记录色谱图。结果表明:与阴性对照样品进行比较,酸性环境制剂有较强破坏作用,产生了更多的杂质。
(3)碱破坏试验
精密称取左亚叶酸钠冻干粉及空白辅料适量(约相当于左亚叶酸50 mg),置于50 mL容量瓶中,加入0.1 mol/L氢氧化钠溶液至刻度,超声使溶解。水浴加热2 h,放冷,滤过,弃去初滤液。精密量取续滤液以流动相稀释制成浓度约为 1 mg/mL(以左亚叶酸计)的样品溶液,精密量取20 μL,注入液相色谱仪,记录色谱图。结果表明:与阴性对照样品进行比较,碱性环境对制剂均有破坏作用,产生了更多的杂质。
(4)热破坏试验
精密称取左亚叶酸钠冻干粉及空白辅料适量(约相当于左亚叶酸50 mg),置于50 mL容量瓶中,加入流动相至刻度,超声使溶解。水浴加热2 h,放冷,滤过,弃去初滤液。精密量取续滤液以流动相稀释制成浓度约为1 mg/mL(以左亚叶酸计)的样品溶液,精密量取20 μL,注入液相色谱仪,记录色谱图。结果表明:与阴性对照样品进行比较,热环境对制剂有一定的破坏作用,产生了更多的杂质。
(5)氧化破坏
精密称取左亚叶酸钠冻干粉及空白辅料适量(约相当于左亚叶酸50 mg),置于50 mL容量瓶中,加入双氧水(30%过氧化氢)至刻度,超声使溶解。水浴加热2 h,放冷,滤过,弃去初滤液。精密量取续滤液以流动相稀释制成浓度约为1 mg/mL(以左亚叶酸计)的样品溶液,精密量取10 μL,注入液相色谱仪,记录色谱图。结果表明:与阴性对照样品进行比较,氧化环境对制剂有很强的破坏作用,产生了更多的杂质。
上述试验结果表明:本品在强光下较稳定,未见明显破坏,在酸破坏、碱破坏、热破坏、氧化破坏条件下均不稳定;但在本色谱条件下各降解杂质峰均能与产品主峰有较好的分离度;表明该色谱条件下可能存在的已知杂质及强降解产生的未知杂质均能被有效检出。证明该方法专属性良好,满足本品有关物质测定需求。
2.4 线性试验
2.4.1 供试品线性试验
精密称取左亚叶酸钠冻干粉适量(约相当于左亚叶酸 125 mg),置于25 mL容量瓶中,加流动相适量,超声使溶解,稀释至刻度,摇匀,滤过,弃去初滤液。取续滤液作为线性试验贮备液。分别精密量取线性试验贮备液1 mL、2 mL、3 mL、4 mL、5 mL、6 mL,置6个25 mL容量瓶中,加流动相稀释至刻度制成0.20~1.20 mg/mL的系列溶液。分别精密量取上述溶液各20 μL,注入液相色谱仪,记录色谱图。结果见表1所示。

表1 供试品线性试验
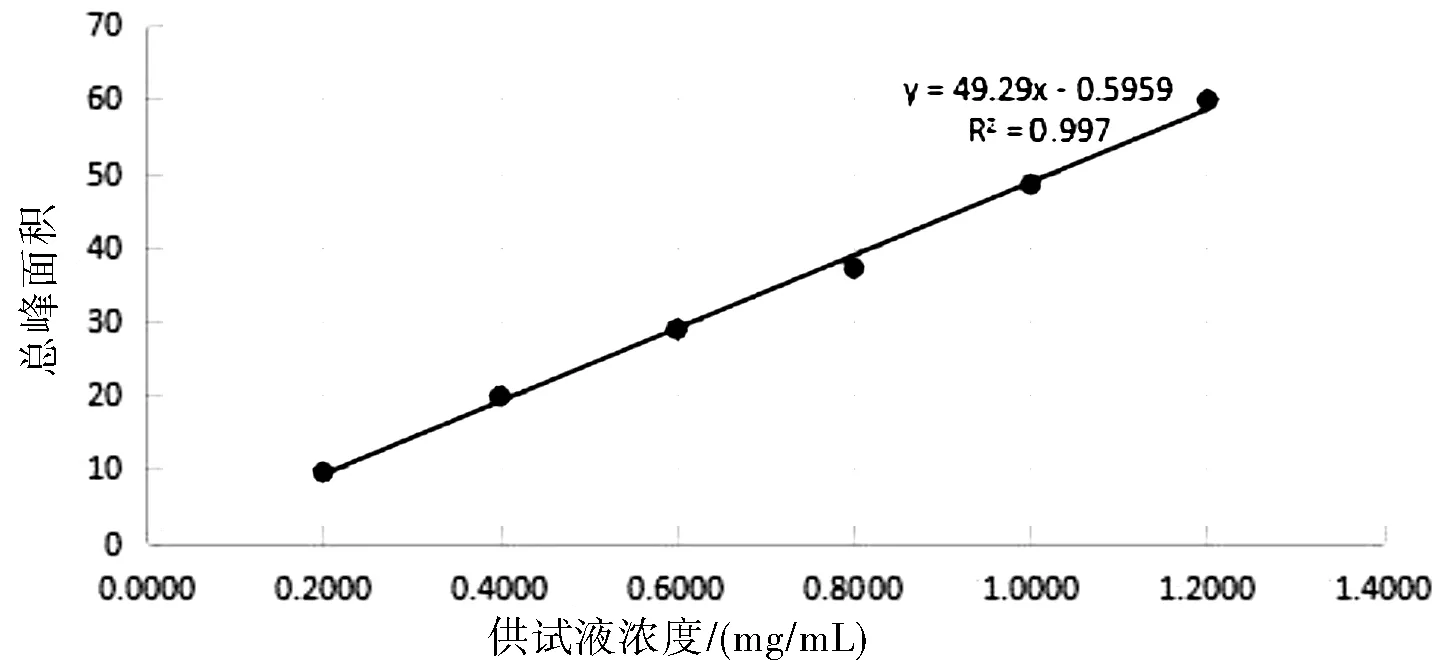
图1 总峰面积线性图
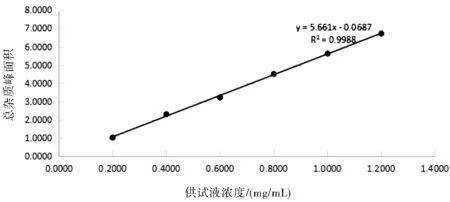
图2 总杂质峰面积线性图
结论:以上数据及图谱表明,供试品溶液浓度在0.20~1.20 mg/mL范围内,总杂质峰面积线性关系良好。
2.4.2 自身对照线性试验
分别精密称取左亚叶酸钠冻干粉适量(约相当于左亚叶酸50 mg),置于50 mL容量瓶中,加流动相适量,超声使溶解,稀释至刻度,摇匀,滤过,弃去初滤液。精密量取续滤液1 mL置于50 mL容量瓶中,加流动相并稀释至刻度,制成浓度为 20 μg/mL的溶液,作为1%自身对照溶液线性试验贮备液。分别精密量取线性试验贮备液2.0 mL、3.0 mL、4.0 mL、5.0 mL、6.0 mL、7.0 mL置于10 mL容量瓶中,加流动相稀释至刻度,摇匀。分别精密量取上述溶液各20 μL,注入液相色谱仪,记录色谱图。结果见表2所示。

表2 1%自身对照溶液线性试验
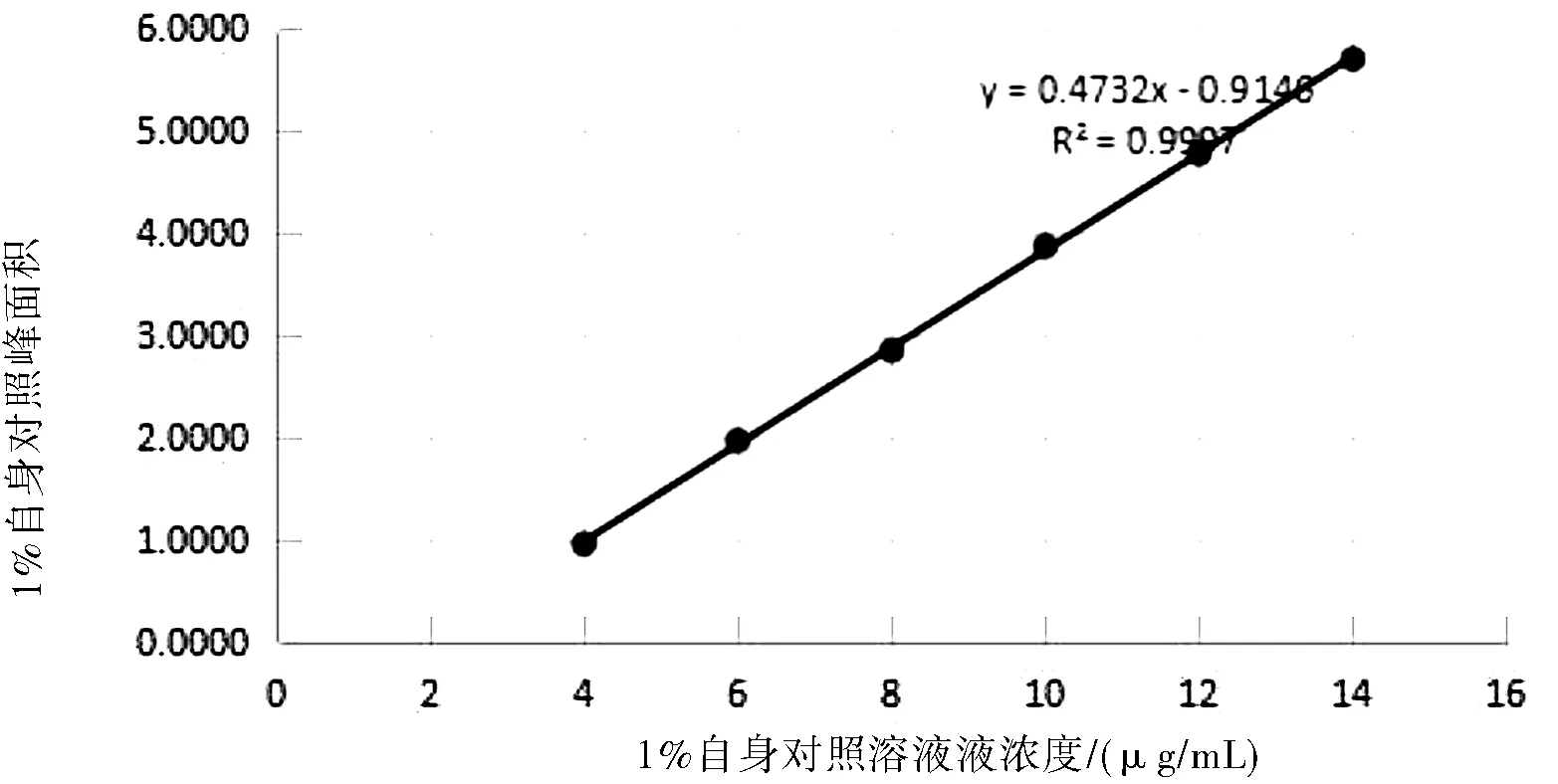
图3 1%自身对照面积线性图
结论:以上数据及图谱表明,1%自身对照溶液浓度在4.0~14.0 μg/mL范围内,主峰面积线性关系良好。
2.5 样品检出限
以1%自身对照溶液线性试验1#溶液为检出限起点样品,分别以流动相稀释10倍、20倍、40倍、80倍,分别精密量取上述溶液20 μL,注入液相色谱仪,记录色谱图,由检出限图谱可知,稀释80倍时,当自身对照溶液浓度为0.0125 μg/mL时,主峰的峰高约为基线噪声比的三倍,即为供试品溶液杂质的最低检出限。
2.6 重复性试验
精密称取左亚叶酸钠冻干粉适量(约相当于左亚叶酸 50 mg),置于50 mL容量瓶中,加入流动相适量,超声使溶解,稀释至刻度,摇匀,滤过,弃去初滤液。取续滤液作为供试品溶液,同法制备6份。分别精密量取上述溶液适量,加流动相稀释制成1 %自身对照溶液。精密量取上述各溶液20 μL,注入液相色谱仪,记录色谱图,结果见表3、表4所示。
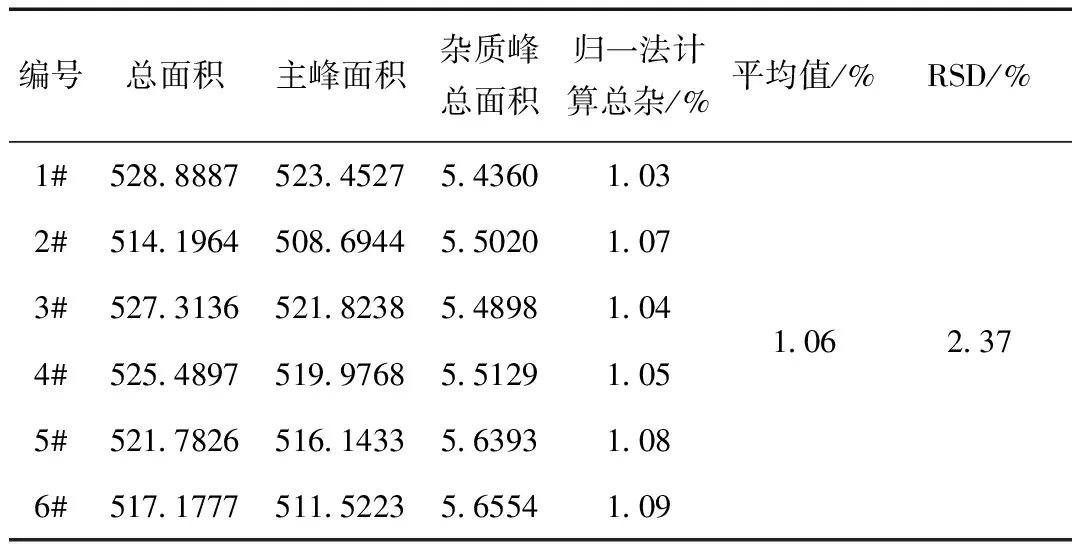
表3 供试品溶液重复性试验结果

表4 1%自身对照溶液重复性试验结果
结论:根据中国药典及中国药品检验标准操作规范的相关规定,以上数据表明,供试品溶液及1%自身对照溶液重复性良好。
2.7 溶液稳定性试验
精密称取左亚叶酸钠冻干粉适量(约相当于左亚叶酸 5 mg),置于25 mL容量瓶中,加入流动相适量,超声使溶解,稀释至刻度,摇匀,滤过,弃去初滤液。取续滤液作为供试品溶液。精密量取供试品溶液1 mL置于100 mL容量瓶中,加流动相稀释至刻度,摇匀,作为自身对照溶液。将上述溶液分别在室温下放置4 h、6 h、8 h、10 h后,精密量取上述各溶液20 μL,注入液相色谱仪,记录色谱图,结果见表5、表6所示。

表5 供试品溶液稳定性试验结果

表6 1%自身对照溶液稳定性试验结果
结论:以上数据表明,供试品溶液及1 %自身对照溶液在室温0~10 h内稳定性良好,总杂质在10 h内虽然略有增加趋势,但0、4、6、8、10 h总杂质RSD仍小于2.0,在偏差可接受范围内,在我们暂定标准中可认为溶液稳定不小于8 h。
2.8 系统适用性试验
色谱柱理论板数:按2.1制供试品溶液,精密量取续滤液20 μL,注入液相色谱仪,记录色谱图至主峰保留时间的3倍以上。
按2.1制备对照溶液(1)和对照液(2)。分别精密量取此液20 μL,注入液相色谱仪,记录色谱图。连续进样,计算相对标准偏差,结果见表7所示。

表7 系统适用性试验结果
结论:液相色谱图及以上数据显示,左亚叶酸峰理论板数 n=11231,主峰与最近杂质峰的分离度为1.76>1.5,对照溶液重复进样相对标准偏差分别为0.29%和0.24%,均小于2.0%。结果表明,色谱条件良好,分离度良好。
2.9 有关物质检查方法及结果
按2.1项制备3批供试品溶液及对照溶液(1)和对照液(2),按2.2确定色谱条件进行测试。三批样品有关物质检查,结果见表8所示。
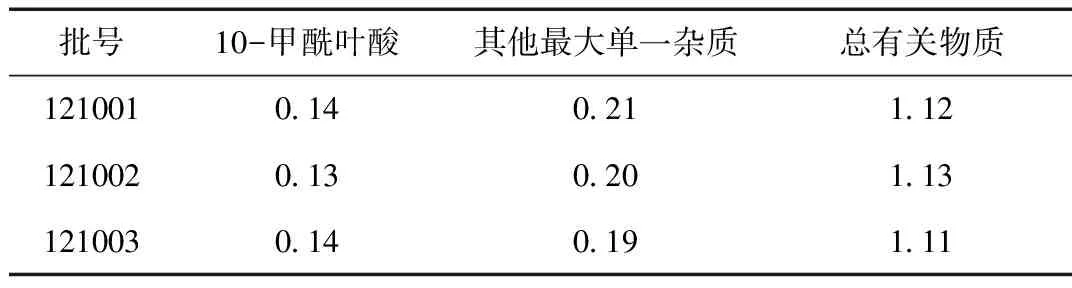
表8 三批产品测定结果
3 结 论
本试验对高效液相色谱法法测定注射用左亚叶酸钠有关物质的测定方法进行了研究,结果表明本试验方法对产品中的杂质均能与主成分峰良好分离,强降解后产生更多杂质也能准确。溶液稳定性试验结果表明样品溶液总杂质在10 h内虽然略有增加趋势,但0、4、6、8、10 h总杂质RSD仍小于2.0,在偏差可接受范围内,暂定溶液稳定时间为8 h。三批样品测定结果本方法用于注射用左亚叶酸钠的有关物质重复性好,结果准确可靠,可用于注射用左亚叶酸钠的有关物质检验。
