含杂质气态CO2环境中X65钢腐蚀行为
2022-07-26刘建新孙建波林学强
孙 冲, 刘建新, 孙建波, 李 晗, 赵 智, 林学强, 王 勇
(1.中国石油大学(华东)材料科学与工程学院,山东青岛 266580; 2.中国石化胜利油田分公司石油工程技术研究院,山东东营 257000; 3.中国石油大港油田公司第六采油厂,河北黄骅 061100)
碳捕集、利用与封存(CCUS)技术是实现CO2深度减排的重要技术途径,是中国中长期应对气候变化、推进低碳发展和保障能源安全的重要战略技术选择[1-2]。在CCUS技术链中,管道是实现大规模CO2输送最经济有效的方法[3],而将捕集的CO2输送到油气田用于驱油(EOR)则是实现CO2规模化利用和地质封存非常有前景的应用方向,可以满足油藏高效开发和环保的双重要求[4]。然而,捕集的CO2中通常含有一定量H2O及腐蚀性杂质(O2、SO2、H2S、NO2等)[5],其与压缩CO2(即高压CO2,如超临界态、密相态、液态或气态[6])一起引入CO2输送和驱注采环节中,将会增加管道及井下工具的腐蚀风险。腐蚀问题是CCUS技术规模化应用过程中不容忽视的重要问题[6-7]。在CCUS过程中各个环节的腐蚀环境存在差异,总体可归为富CO2相环境和富H2O相环境[4,8]两类。前者CO2为主体,H2O和杂质溶于CO2,该类环境主要存在于CO2输送或注入环节;后者H2O为主体,CO2、杂质及各种矿物离子等溶于H2O,该类环境主要出现在CO2注入或油气生产环节。这也导致碳钢管材在两种环境中的腐蚀行为存在一定差异。通常,由于侵蚀性液相量大且相对稳定,碳钢管材在富H2O相中比在富CO2相中面临更严重的腐蚀[9-11]。与传统油气田CO2环境中的CO2腐蚀机制不同,在含杂质CO2环境中除了CO2腐蚀外,O2、SO2、NO2或H2S等杂质可以不同程度地控制腐蚀过程[12-14]。而多种杂质共存时,杂质之间还可以发生复杂的交互作用,生成额外的腐蚀性物质,如H2SO4、S、HNO3、NH4+等,进而协同加剧碳钢的腐蚀,导致腐蚀机制更加复杂[15-18]。与富CO2相环境相比,在富H2O相中杂质能够带来更强的腐蚀促进效应[12,19]。然而,在含多种杂质(O2、SO2和H2S)的超临界CO2环境中碳钢管材在富CO2相中的腐蚀更为严重,且杂质对腐蚀的影响比其在富H2O相中更为突出[20]。然而,对于含杂质CO2流体的腐蚀研究主要集中于超临界态或密相态CO2环境,含杂质气态CO2环境的腐蚀研究较少。而在CCUS应用过程中除了含杂质超临界/密相态CO2腐蚀环境,碳钢管材也会面临含杂质的气态CO2腐蚀环境,其腐蚀行为以及杂质对腐蚀的影响必然会因CO2相态的差异而有所不同[21-22]。尤其是中国目前已有的CO2管道均为气态CO2输送管道[23]。考虑中国CCUS技术的应用现状,研究含杂质气态CO2环境中碳钢管材的腐蚀规律及机制,更具有现实意义。笔者针对含O2、SO2和H2S杂质的气态CO2腐蚀环境,通过腐蚀模拟实验、表面分析技术和水化学模拟计算及腐蚀预测等手段,对比研究X65钢在富CO2相和富H2O相环境中腐蚀行为的差异,探讨杂质对X65钢腐蚀的影响机制。
1 实验方法
实验材料选用商用X65管线钢,其化学成分(质量分数)为:C(0.06),Si(0.288),Mn(1.52),P(0.012),S(0.003),Mo(0.178),Cr(0.048),Ni(0.008),Al(0.057),Cu(0.007),V(0.031),Fe余量。试样尺寸为40 mm×15 mm×3 mm,其表面用砂纸逐级打磨至1000#,用去离子水冲洗、乙醇脱水、丙酮脱脂、冷风吹干。采用电子天平(精度为0.000 1g)称量试样的初始质量。实验介质为质量分数3.5%的NaCl溶液,采用高纯N2除氧至少12 h。
腐蚀模拟实验在3 L容积的高温高压反应釜中进行,实验装置见图1。实验条件如下:温度为50 ℃,CO2压力为5 MPa,O2、H2S和SO2杂质体积分数均为1 000×10-6。每组实验设置4个平行试样,其中3个试样用于计算腐蚀速率,1个试样用于腐蚀膜微观分析。如图1所示,一组试样放置于富CO2相(水饱和含杂质气态CO2相),模拟气态CO2输送或注入环境;另一组试样放置于富H2O相(含杂质CO2饱和水溶液),模拟气态CO2注入或驱油环境。实验前,将试样悬挂在聚四氟乙烯夹具上,向反应釜中加入1 L除氧的NaCl溶液。然后,向关闭的反应釜中通入CO22 h,以除去安装过程中釜内残留的空气。首先升温至50 ℃,然后分别向反应釜中加入O2、H2S和SO2至所需浓度,最后加入CO2至设定压力。腐蚀时间分别为1.5、6、24、48和72 h。所有实验均在静态条件下进行。
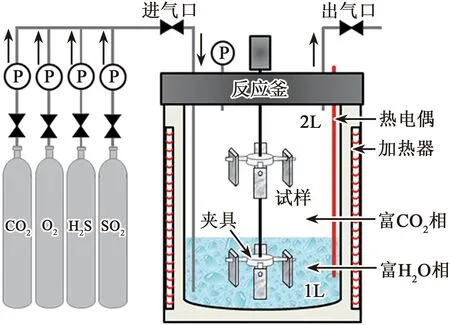
图1 腐蚀实验装置示意图
实验结束后,取出试样。将暴露于富CO2相中腐蚀后的试样置于真空干燥皿中自然脱水,将暴露于富H2O相中腐蚀后的试样用去离子水清洗、乙醇脱水、冷风吹干。用0.5 L盐酸(密度为1.19 g/mL)和3.5 g六次甲基四胺(C6H12N4)及去离子水配置的1 L溶液去除试样表面腐蚀产物[24],干燥后称重。采用失重法计算试样的腐蚀速率。腐蚀速率为
(1)
式中,v为腐蚀速率,mm/a(腐蚀速率为3个平行试样速率的平均值);ΔW为试样腐蚀前后的失重量,g;S为试样表面积,cm2;ρ为试样密度,取7.85 g/cm3;t为腐蚀时间,h。
采用扫描电子显微镜(SEM)观察腐蚀膜的表面形貌,用能谱仪(EDS)分析腐蚀膜的元素组成。再截取部分试样,用环氧树脂封装,用砂纸打磨至2000#,观察试样截面背散射电子像,并分析截面腐蚀膜的元素分布。采用X射线衍射仪(XRD)测定腐蚀产物的物相组成,其工作条件为44 kV,44 mA,Cu靶。用X射线光电子能谱(XPS)分析腐蚀产物的化学价态(Mg靶,光子能量为1 253.6 eV),本文中参照C 1s峰结合能284.8 eV对XPS图谱进行荷电校正。
为了探明富CO2相和富H2O相环境中液相的化学特性及其对腐蚀的影响,采用OLI Analyzer Studio软件的Stream Analyzer和Corrosion Analyzer模块进行水化学热力学计算及腐蚀预测。根据腐蚀实验条件,确定用于模拟计算的腐蚀介质组成。如图1所示,3 L反应釜中腐蚀介质包括2 L含杂质CO2和1 L 3.5 %NaCl溶液。在5 MPa和50 ℃条件下CO2密度为104.90 kg/m3[25],计算可得反应釜中CO2质量为210 g。本研究中杂质质量分数为1 000×10-6,对应质量为0.210 6 g。因此,富H2O相环境的介质组成为965 g H2O,35 g NaCl,210 g CO2,0.210 6 g O2,0.210 6 g SO2和0.210 6 g H2S。由于很难测量富CO2相环境中实际沉积在钢表面的液相量,假设0.1 g H2O沉积在单位面积(1 cm2)的钢表面上,即假设富CO2相环境中可以在钢表面形成厚度为1 mm的均匀液膜。因此富CO2相环境的介质组成可确定为0.1 g H2O,210 g CO2,0.210 6 g O2,0.210 6 g SO2和0.210 6 g H2S。
2 实验结果与讨论
2.1 腐蚀速率
图2为含杂质的气态CO2环境中X65钢腐蚀速率随腐蚀时间变化的曲线。在腐蚀起始1.5 h内,暴露于富CO2相和富H2O相中的X65钢腐蚀速率分别高达9.23和8.34 mm/a。虽然随着腐蚀时间延长,腐蚀速率显著降低;但是腐蚀72 h后,在两种相环境中的腐蚀速率仍处于较高水平,分别为0.96和2.22 mm/a。根据NACE标准RP0775-2005中基于均匀腐蚀速率对碳钢腐蚀程度的分类[26],在这两种环境的测试周期内(72 h)X65钢的腐蚀速率均远高于0.25 mm/a,可归类为最高等级的严重腐蚀程度。由此可见,在含杂质的气态CO2环境中腐蚀初期碳钢管材遭受严重的腐蚀。
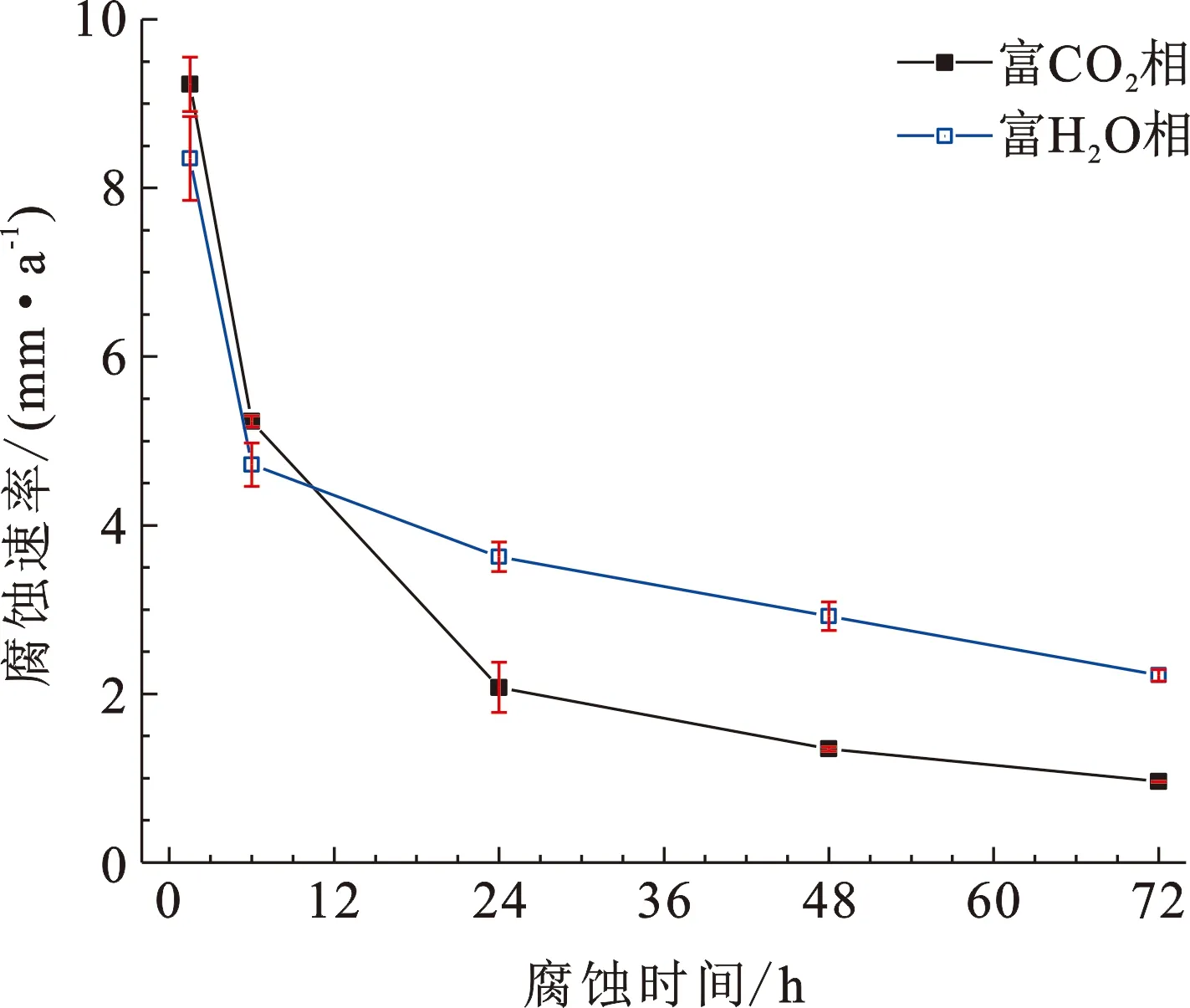
图2 在富CO2相和富H2O相中不同腐蚀时间后X65钢的腐蚀速率
图3中对比了相同杂质和温度条件下在气态CO2环境(5 MPa)与前期报道[20]的超临界CO2环境(8 MPa)中X65钢腐蚀速率的差异。由图3可见,在气态CO2环境中X65钢的腐蚀速率明显低于超临界态CO2环境。尤其是在富CO2相中,CO2相态的变化(即压力的变化)对腐蚀速率的影响更为显著(图3(a))。值得注意的是,在超临界态CO2环境条件下72 h测试周期内,X65钢在富CO2相中的腐蚀速率始终高于其在富H2O相中的腐蚀速率[20]。而在气态CO2环境条件下腐蚀前6 h内,X65钢在富CO2相中的腐蚀速率高于其在富H2O相中的腐蚀速率,腐蚀24 h后,X65钢在前者中的腐蚀速率低于后者(图2)。基于不同均匀时间段(24 h)内X65钢在富H2O相中失重量WH2O和富CO2相中失重量WCO2,计算可得二者失重比值WH2O/WCO2,如表1所示。随着腐蚀时间延长,WH2O/WCO2不断增加,即随着腐蚀的进行,二者的实时溶解速率差距不断增大。可见与超临界态CO2环境中的腐蚀情况不同,在气态CO2环境中X65钢在富H2O相中腐蚀更为严重。

图3 含相同杂质的气态CO2环境与超临界态CO2环境中X65钢的腐蚀速率对比

表1 不同腐蚀时间段内X65钢在富H2O相中失重量与在富CO2相中失重量比值(WH2O/WCO2)
2.2 水化学分析
与富H2O相中裸钢直接与大量液相介质接触而发生腐蚀不同,在富CO2相中H2O首先从CO2相中析出,然后以液相薄膜或液滴形式沉积在裸钢表面上[17,27],同时CO2及O2、H2S和SO2等杂质溶于液相中,进而引起钢的腐蚀。相应地,在富CO2相和富H2O相中腐蚀初期X65钢的腐蚀速率差异与杂质及CO2引起的液相化学特性变化密切相关。
两种环境的液相化学分析计算结果,如表2所示。相比富CO2相,富H2O相中的液相含有更多腐蚀性物质。然而,前者液相中主要腐蚀性物质(H+、CO2(aq)、SO2(aq)、H2S(aq)、O2(aq)等)的浓度更高,而且pH值比后者更低。显然,当X65裸钢暴露于富CO2相时,会面临更加恶劣的液相腐蚀环境。图4为基于液相化学环境预测的极化曲线结果(图中电位相对于SHE)。由图4可见,在两种相环境中X65钢表面发生的电极反应相同。然而,各种杂质对二者阴极过程的影响程度不同,导致二者自腐蚀电流密度存在差异。在富CO2相中自腐蚀电流密度明显高于富H2O相,说明在腐蚀起始阶段X65裸钢在富CO2相薄液膜环境中具有更高的腐蚀速率,这与腐蚀模拟实验结果相吻合(图2)。然而,在富H2O相中由于液相量大,其化学环境相对稳定。在富CO2相中由于液相量少,且处于不断消耗与再沉积的动态过程(干湿交替)以及腐蚀产物形成等因素,导致钢表面的液相化学环境不稳定。这可能导致在富CO2相腐蚀中后期(24~72 h)X65钢的腐蚀速率低于其在富H2O相中的腐蚀速率(图2)。
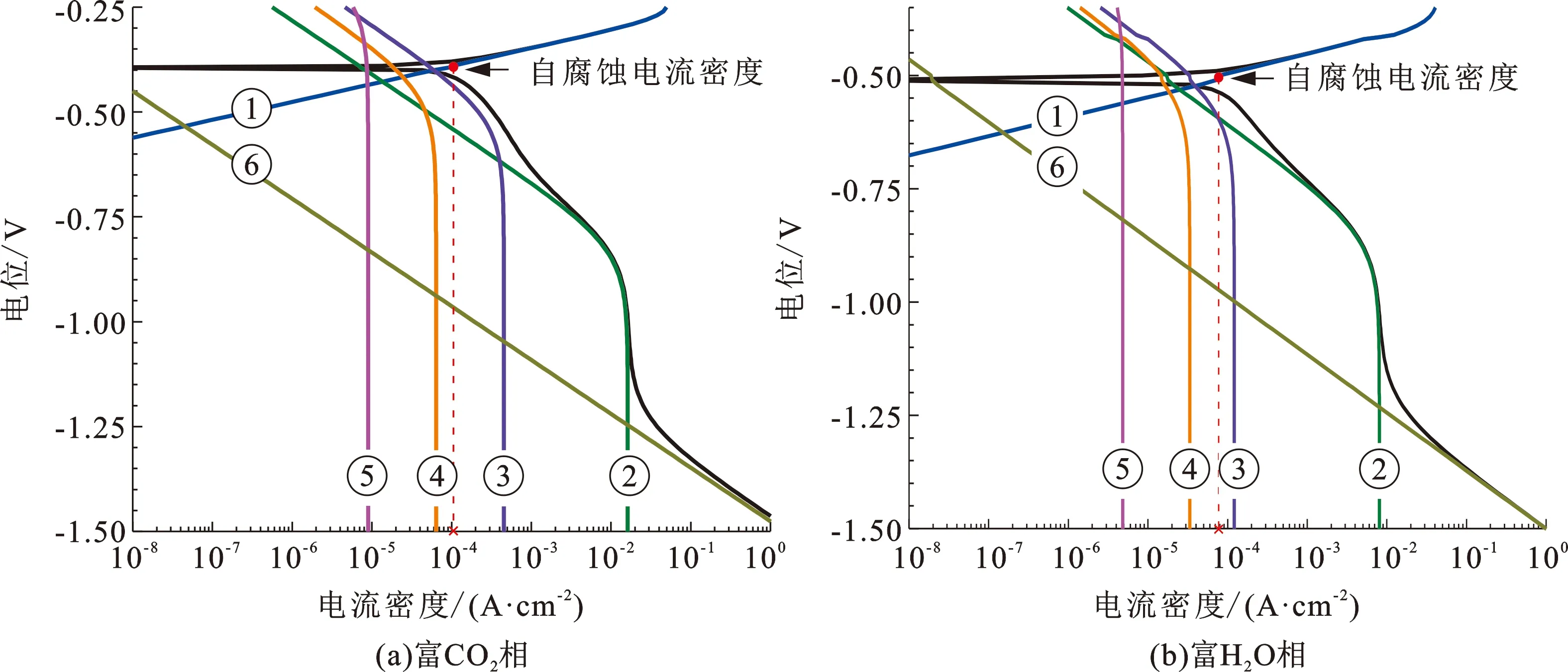
图4 在5 MPa,50 ℃时CO2相和富H2O相中Fe的极化曲线及主要的阳极和阴极反应(OLI Analyzer Studio软件计算)

表2 在5 MPa,50 ℃时富CO2相和富H2O相中液相的主要化学物质和pH值(OLI Analyzer Studio软件计算)
2.3 腐蚀产物膜表征
2.3.1 宏观形貌观察
图5为在含杂质的气态CO2环境中不同腐蚀时间后X65钢表面的宏观形貌。类似于富H2O相环境(图5(b)),在富CO2相中腐蚀1.5 h后灰黑色腐蚀产物已经覆盖整个钢表面(图5(a))。这间接地说明在腐蚀最初阶段富CO2相中的水可以快速析出,并在整个钢表面凝结形成腐蚀性液相薄膜,进而引起整个表面的腐蚀;然而,实验结束后观察取出的试样表面,并未发现肉眼可见的液相。说明伴随着腐蚀发生与腐蚀产物的沉积,钢表面形成的液相也在不断消耗,并被腐蚀产物饱和。这也证实在富CO2相中X65钢表面形成的液相环境是不稳定的,同时液相的腐蚀性也会被削弱,导致后续腐蚀过程中钢溶解速率大大降低。

图5 在5 MPa,50 ℃时富CO2相和富H2O相中不同腐蚀时间后X65钢表面宏观形貌
随着腐蚀时间延长,暴露于富CO2相中的腐蚀膜宏观形貌特征发生显著变化,腐蚀膜变得非常蓬松,轻触即可脱落,其颜色也逐渐转变为黄棕色(图5(a))。相比之下,暴露于富H2O相中的腐蚀膜宏观特征变化比较温和,但在腐蚀72 h后,其表面也可观察到一些黄棕色产物(图5(b))。上述现象与前期报道[20]的相同条件下X65钢在超临界态CO2环境中腐蚀膜宏观特征的变化规律基本类似。这可能意味着在含O2、H2S、SO2的气态CO2环境中X65钢腐蚀膜的演化不受CO2相态变化的影响。
2.3.2 SEM形貌观察与EDS分析
图6为不同腐蚀时间后X65钢腐蚀膜的SEM表面形貌及截面背散射电子像,其中1#~7#标记处腐蚀产物的EDS分析结果,如表3所示。在两种环境中腐蚀1.5 h后,X65钢表面均形成一层胶泥态龟裂的腐蚀膜,局部可观察到一些颗粒状产物。EDS分析结果显示腐蚀产物主要包含Fe、S和O元素(表3),说明腐蚀产物由铁的氧化物或硫化物组成。
随着腐蚀进行,腐蚀膜表面微观形态、元素组成及结构特征均发生明显变化。如图6(a)所示,在富CO2相中腐蚀24 和72 h后,腐蚀膜均呈双层膜结构。EDS线扫描分析表明内层主要含有Fe和O元素,外层主要含有Fe、S和O元素。相比腐蚀24 h后的外层膜,腐蚀72 h后的外层膜更加疏松,且膜中O/S含量比显著增加,即S元素含量大大降低(表3)。这说明随着腐蚀时间延长,暴露于富CO2相中的腐蚀膜表层很可能发生进一步氧化,导致膜中硫相关产物含量大大降低。
与富CO2相类似,在富H2O相中腐蚀24 h和72 h后,X65钢表面也形成双层结构膜,内层膜O含量较高,而外层中S含量较高(图6(b))。此外,在腐蚀72 h后的外层膜表面还可以观察到一些蓬松的腐蚀产物(图6(b)中7#标记处,即宏观形貌观察到的黄棕色产物),这些产物中O/S含量比明显高于外层膜(图6(b)中6#标记处),说明其氧化物含量较高。相比腐蚀24 h后的外层膜,腐蚀72 h后的外层膜的形态及厚度变化不大,但是膜中O/S含量比有所升高(即S含量降低),如表3所示。总体上,在24~72 h腐蚀时间内,在富H2O相中X65钢腐蚀膜特性的变化不如其在富CO2相中显著。
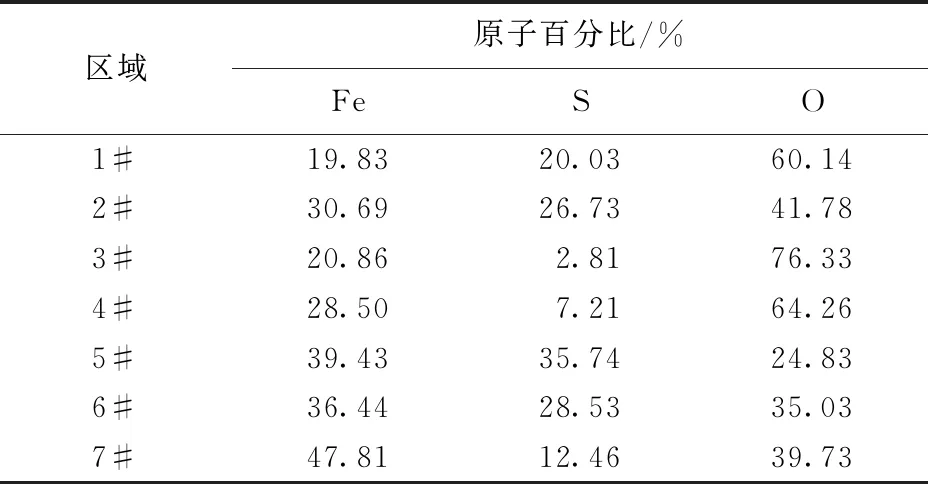
表3 X65钢腐蚀膜表层EDS分析(图6中1#~7#标记区域)
2.3.3 XRD和XPS分析
图7为不同腐蚀时间后X65钢腐蚀膜的XRD图谱。由图7可见,在两种环境中腐蚀1.5 h后,腐蚀膜的XRD图谱中只检测到FeS产物的衍射峰(Fe的衍射峰来源于钢基体,这是由于腐蚀膜较薄,低于XRD衍射深度所致)。然而,EDS分析表明腐蚀膜中含有大量O(表3),XRD图谱中却未检测出含氧产物的衍射峰。这说明腐蚀起始阶段的强酸性液相环境不利于晶态含氧产物的形成,其在腐蚀膜中主要以非晶态形式存在。而FeS在强酸性环境中具有更高的化学稳定性[28],其主要以晶态形式存在,因而XRD图谱中主要检测到FeS的衍射峰。在富CO2相中腐蚀72 h后,XRD结果表明腐蚀膜主要由FeCO3、FeSO4·xH2O、FeS、S和FeOOH组成。这进一步证实前文中的推论,富CO2相中形成的液相环境是不稳定的。随着腐蚀的进行,由于液相环境的变化含氧产物能够以晶体形态析出。然而在富H2O相中腐蚀72 h后,XRD图谱并未发生明显变化,主要显示FeS的衍射峰,说明含氧产物仍处于非晶态,这也间接证实富H2O相中的液相环境更加稳定。
显然,在富H2O相中形成的大部分产物呈非晶态,通过XRD方法不能获取更多腐蚀膜组分的信息。为了明确富H2O相中腐蚀产物的组成,采用XPS进一步分析了腐蚀产物的化学状态,并与富CO2相中腐蚀产物对比,结果如图8所示。由图8可见,两种环境中腐蚀产物的Fe 2p、O 1s、S 2p和C 1s高分辨XPS图谱类似,XPS特征峰出现在相近的结合能处,这说明腐蚀产物具有相同的化学状态。由图8(a)可见,Fe 2p3/2和Fe 2p1/2主峰分别位于711.2 eV和724.9 eV结合能处,表明Fe处于氧化态(Fe2+或Fe3+)[29-30]。由于铁的相关化合物具有相近的结合能,仅根据Fe 2p谱难以确定具体的产物类型。因此本文中主要根据O 1s、S 2p和C 1s图谱分峰拟合结果确定腐蚀产物。如图8(b)所示,O 1s峰可分解为3个特征峰,位于530.0、531.5和532.4 eV结合能处,分别对应于氧化物/羟基氧化物、碳酸盐和硫酸盐[30-32]。根据S 2p图谱分峰结果(图8(c)),在161.5、163.6和168.7 eV处存在3个S 2p3/2特征峰,表明硫在腐蚀产物中分别以硫化物、单质硫和硫酸盐的形式存在[30-31,33]。此外,C 1s图谱(图8(d))在289.4 eV处的特征峰与碳酸盐相对应[29],而在284.8和286.3 eV处的特征峰源自外来碳。综合考虑各元素的化学状态,可以确定在富CO2相中腐蚀72 h后腐蚀产物的主要成分为FeOOH、FeCO3、FeSO4、FeS和S,这与图7(a)中XRD分析结果相吻合。鉴于富H2O相和富CO2相中腐蚀产物具有相同的化学状态,可以推断富H2O相中腐蚀产物主要包含FeOOH、FeCO3、FeSO4、FeS和S。

图7 在富CO2相和富H2O相中腐蚀不同时间后X65钢腐蚀膜的XRD图谱
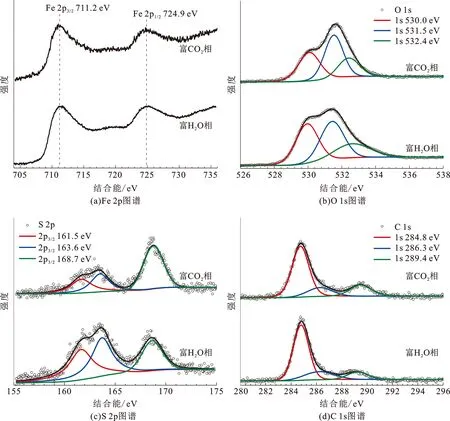
图8 在富CO2相和富H2O相中腐蚀72 h后X65钢腐蚀膜中不同元素的高分辨XPS图谱
2.4 腐蚀机制
在含O2、H2S和SO2杂质的气态CO2环境中腐蚀72 h后,X65钢在富CO2相和富H2O相中腐蚀膜物相组成相同,主要为FeCO3、FeSO4、FeS、S和FeOOH,说明在两种环境中X65钢的基本腐蚀反应及成膜机制类似。CO2和H2S可以直接参与腐蚀过程,形成特征产物FeCO3和FeS。而O2和SO2共存时,二者反应可以生成H2SO4[18],表示为

因此,除了CO2和H2S腐蚀,X65钢也受到H2SO4腐蚀作用,生成特征产物FeSO4。此外,H2S与SO2或O2之间也可以发生化学反应,导致单质S的形成[18,34-35],表示为
基于本文的研究结果,无法确定生成的单质S是否参与了X65钢的腐蚀过程;但相关研究已表明在含O2、SO2、H2S等杂质的CO2环境中杂质交互作用形成的单质S能够引起碳钢的元素硫腐蚀,这是含多种杂质CO2环境中碳钢腐蚀加剧的重要原因之一[34-35]。
除了上述腐蚀过程以外,由于强氧化性杂质O2的存在,与介质直接接触的表层腐蚀产物容易被氧化[12,36-37],表示为
可见,腐蚀膜的氧化不仅导致FeOOH或S形成,还会造成侵蚀性酸性物质的循环再生(如H2SO4)[12],其会继续对钢基体造成腐蚀,这可能是X65钢在腐蚀后期仍具有较高腐蚀速率的原因之一(图2)。腐蚀72 h后腐蚀膜颜色的显著变化(图5)、表层膜O/S含量比的明显增加(表3)以及XRD图谱中FeS衍射峰数量的明显减少(图7(a))均证实腐蚀膜的氧化现象,尤其是在富CO2相中膜的氧化现象更为突出。一方面在相同条件下绝大部分O2分布于富CO2相(图9),另一方面相比富H2O相中的大体量液相介质,富CO2相中形成的薄液膜更有利于腐蚀性物质(如O2)的补充和传输[38-40]。因而,暴露于富CO2相中的腐蚀膜可以与充足的O2接触,容易发生氧化。相比之下,在相同条件下进入富H2O相的O2量非常低。如图9所示,1 000×10-6O2中只有约10×10-6溶于液相,即在与富CO2相环境相同的条件下,只有非常有限的O2参与表面膜的氧化。相应地,在富H2O相中腐蚀膜随时间的变化相对温和且缓慢(图5(b)和图6(b))。分析表明,在含杂质的气态CO2环境中,腐蚀反应、杂质交互作用以及腐蚀膜氧化等共同影响X65钢腐蚀膜的形成。
对图4中阳极和阴极极化曲线进一步分析,可见在薄液膜和大体量液相中X65钢的腐蚀电化学机制相同,这与腐蚀膜表面分析得出的结论一致。如图4所示,阳极过程只发生了Fe氧化为Fe2+的反应(图4中①)。这证明腐蚀膜中3价Fe产物(如FeOOH)主要源自2价Fe产物的氧化,而不是直接的腐蚀反应,如氧腐蚀反应。对比阴极反应可见,O2的阴极去极化作用(图4中⑤)在阴极过程中非常弱,说明在测试条件下X65钢发生O2腐蚀的可能性很低。阴极过程由H+(源自CO2、SO2和H2S水解)、H2CO3和H2S的还原主导。显然,3种杂质中O2对阴极反应的直接影响远不及SO2和H2S,这与其在CO2相和液相中的含量密切相关。由图9可见,SO2和H2S在液相中的溶解度远高于O2。虽然O2对阴极过程的影响很小,但是O2可以与SO2反应生成H2SO4,可为腐蚀反应提供更多H+。因此O2可以间接地促进阴极反应过程,其在腐蚀中的影响不容忽视。相比富H2O相中的阴极反应,富CO2相中与CO2、SO2(主要贡献H+)及H2S相关的阴极析氢反应的电流密度均明显增加(图4中②~④),尤其是H+还原反应③。这提高了阴极电流密度,进而导致X65钢腐蚀电流密度增加。因此,在富CO2相中X65钢具有更高的初始腐蚀速率(图2)。由此可见,X65钢在两种环境中初始腐蚀行为主要由阴极过程控制,而腐蚀速率的差异主要与CO2和杂质引起阴极过程的变化有关;而随着腐蚀进行,受薄液膜环境不稳定性(干湿交替)及腐蚀产物形成等影响,在富CO2相中X65钢具有更低的腐蚀速率(图2)。

图9 在5 MPa和50 ℃条件下1 000×10-6不同杂质在CO2相和液相中的质量分数(OLI Analyzer Studio软件计算)
3 结 论
(1)在含O2、H2S和SO2杂质的气态CO2环境中,X65钢在富CO2相和富H2O相中均发生严重腐蚀;由于富CO2相的初始液相化学环境比富H2O相更加苛刻,X65裸钢在富CO2相中具有更高的初始腐蚀速率;而从长期腐蚀上看,由于富H2O相的液相化学环境更稳定,X65钢在富H2O相中发生更为严重的腐蚀。
(2)在富CO2相和富H2O相中X65钢的腐蚀反应机制及成膜机制相同,腐蚀过程由CO2和杂质的直接腐蚀作用、杂质的交互作用及腐蚀膜的氧化作用共同控制,腐蚀膜主要由FeCO3、FeSO4、FeS、S和FeOOH组成。
(3)在气态CO2环境中O2、H2S和SO2杂质通过恶化液相化学环境、促进阴极反应过程及改变腐蚀膜特性显著改变X65钢的CO2腐蚀过程,但在富CO2相和富H2O相中其影响程度不同,导致X65钢在两种环境中的腐蚀速率及腐蚀膜沉积状态存在差异。
